3、碳正离子的内能:
叔<仲<伯<甲基<一氯甲基<二氯甲基<三氯甲基
2、C=O(醛酮)的亲核加成反应活性:
CO(Cl)2 > HCHO > RCHO > RCOR
1、电子效应
⑴诱导效应(±I):
诱导效应基于原子的电负性,它影响物种(分子、离子、自由基、过渡态等)的内能和电子云密度的分布,从而也影响反应活性和选择性。如:
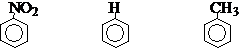
电子云密度: < 1 =1 > 1
硝化反应相对速度: < 1 =1 > 1
取代基的性质: 吸电子基(-I) 供电子基(+I)
诱导效应对物种和反应的影响规律如下:
⑴+I可以增大体系的电子云密度,从而有利于亲电反应,不利于亲核反应;
⑵+I可以分散体系的正电荷,使正离子的内能降低,相反,+I可以使体系的负电核更加集中,导致负离子内能升高。
⑶-I的效应完全与+I相反。
例如:1、烯烃的亲电取代反应活性:
四取代乙烯>三取代乙烯>二取代乙烯>一取代乙烯
>乙烯>氯乙烯>二氯乙烯>三氯乙烯>四氯乙烯
2、基本反应机理的种类:
结合以上两种分类方式,可以把基本有机反应机理分为12种。它们是:
自由基取代、自由基加成、自由基消除、自由基重排
亲 电 取代、亲 电 加成、亲 电 消除、亲 电 重排
亲 核 取代、亲 核 加成、亲 核 消除、亲 核 重排
其中比较重要并常见的有8种,它们是:
自由基取代(烷烃)、自由基加成(烯烃)、亲电取代(芳烃)、亲电加成(烯烃)、亲核取代(卤代烃、醇、酯)、亲核加成(醛酮)、亲核消除(卤代烃、醇)、亲核重排(大部分著名的重排反应)。
反应机理从电子转移角度有机反应过程,更容易离解反应的本质。
1、反应机理的分类:
(1)按产物与反应物之间的关系分为四类
取代反应:分子中的某个原子或原子团被其它原子或原子团取代。

加成反应:分子中的p键断裂,两侧各加上一个原子或原子团。
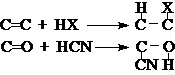
消除反应:相邻原子上各去掉一个原子或原子团生成p键。

重排反应:分子碳干发生改变的反应。
(2)按共价键的断裂方式分为三类:
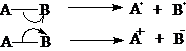
均裂属自由基(游离基)反应,如果存在中间体,中间体必定是自由基。
异裂属离子型反应,如果存在中间体,中间体必定是离子。
离子型反应分为:亲电反应:反应物提供电子,试剂接受电子;
亲核反应:试剂提供电子,反应物接受电子。
2、过渡态理论涉及的重要概念:
①过渡态:曲线上的极大值(峰),反应进程中的动态点,寿命为零
(半衰期T1/2=0),不可观测,是一种理论假设。
②中间体:曲线上的极小值(谷),反应进程中的中间站,寿命大于零(半衰期T1/2>0),是真实存在的粒子,可以观测,其中比较稳定的甚至可以分离。有机反应常见的中间体有:、自由基、碳正离子、碳负离子、卡宾、苯炔等。
③能垒△E和反应速度V的关系:
对于协同式反应,△E是过渡态与反应物之间的能量差,△E越大,反应速度越慢(或者说反应的活性越小),反之△E越小,反应速度越快(反应活性越大)。
对于非协同式反应,由于反应分步进行,所以有两个过渡态、两个能垒。第一过渡态与反应物之间的能量差为第一能垒△E1,第二过渡态与中间体之间的能量差为第二能垒△E2,当△E1 >△E2时,第一步反应为慢反应,整个反应的反应速度取决于第一步;当△E1<△E2时,第二步反应为慢反应,整个反应的反应速度取决于第二步。
利用同位素效应可以确定慢反应步骤,如:C-D键断裂需要的能量比C-H键大,反映在速度上关系为2 : 3。例如:
C6D6硝化速度与C6H6相同,而C6D6的磺化速度却只有C6H6的2/3。怎样认识这种差别呢?
这是因为硝化和磺化都是苯环上的亲电取代反应,其基本反应历程可以表示如下:
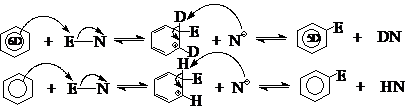
反应分两步进行,C-H或C-D键的断裂发生在第2步。如果对于硝化,第(1)步慢,整个反应的速度取决于第(1)步,而在这一步中,并不涉及C-H或C-D键的断裂,所以苯和氘代苯的硝化速度相同,不表现出同位素效应;而表现出同位素效应的磺化反应,第(2)步为慢反应,整个反应速度取决于第(2)步,而这一步涉及到C-H或C-D键的断裂,所以必定表现出同位素效应。
④热焓△H与反应的热效应
热焓:产物与反应物之间的能量差称为热焓:E产物-E反应物 = △H
吸热反应:△H为正值; 放热反应:△H为负值。
⑤反应分子数和反应级数
反应的分子数:属于反应机理的范畴,是指在决定反应速度的慢反应一步中,组成过渡态的分子个数,它只能是整数。目前,只发现有单分子反应(1)和双分子反应(2)两种模式,更高分子数的反应从几率上考虑是不可能的。设想:3个或更多个分子在空间某一点、同时碰撞在适当的位置(包括分子的活性中心和空间方向等),在几率上可以认为等于零。
反应的级数:属动力学的范畴,是指在反应速率的方程式中,各物种浓度方次之和。虽然理论上也有反应级数的概念,但其实际意义并不大;在实践中,反应级数主要是通过实验去测定,根据实验的具体情况(物料浓度的比例,反应条件和测定的准确性等),它既可能是整数,也可能是分数或小数。
⑥哈蒙德假设 (Hammond Postulate)
哈蒙德假设是针对过渡态理论的一个重要概念。由于反应的过渡态是一种理论上的假设,不可能观测,所以过渡态的几何形状是“不可知的”。由于过渡态的“不可知
性”,导致能垒△E的不可测性(不能直接测量,当然可以通过平衡常数间接获得-见热力学第二定律)。为了了解过渡态的几何状态和能量高低, Hammond提出,在反应过程中,体系(分子、离子或过渡态)的几何状态与其能量具有平行关系,或者说,在反应过程中,过渡态的几何状态与能量接近的粒子(包括反应物、产物、中间体)相似;哈蒙德假设为我们提供了一个考虑过渡态的几何状态和能量高低的方法和途径。
这里可以分为两种情况:
⑴对于协同式反应,过渡态的几何结构介于反应物和产物双方之间,与能量较接近的一方更相似。因此,在吸热反应中,过渡态与反应物物相似,在放热反应中,过渡态与产物相似。
⑵对于非协同式反应,过渡态在能量上更接近于中间体,因此过渡态在几何结构上与中间体相似。因此,我们可以认为:过渡态与中间体具有平行关系。即:中间体的能量越低(越稳定),过渡态的能量也越低,相应的反应的能垒△E就越小,反应速度越快(反应活性越大)。
对于非协同反应,通过中间体的内能来比较反应的活性,是讨论化学问题常用的方法。哈蒙德假设提出之后,探索和研究反应的中间体成为反应理论研究的一种重要手段和领域,至今仍然十分活跃。如:
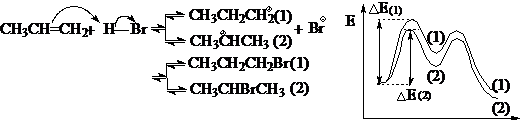
由于中间体(2)的内能较低,所以对应的过渡态内能较低,相应的能垒较小,反应速度较快,经过中间体(2)生产的产物(2)较多,所以反应的主要产物为产物(2)-马可夫尼科夫规则。
⑦微观可逆性和宏观可逆性
从微观上说,任何反应都是可逆反应,而且正反应和逆反应的途径相同,能量曲线完全一样。但在宏观上,只有正、逆反应的能垒差别(△E正-△E逆)小到一定程度,反应成为宏观不可逆反应。当然,由于沉淀、气体和其它因素导致一种或几种产物脱离反应体系,使逆反应失去发生的条件,则是另一种情况(平衡移动原理)。
⑧反应的动力学控制和热力学控制-动力学产物和热力学产物
应当记住:
①首先动力学控制和热力学控制只适用于宏观可逆反应;
②在多数情况下,动力学和热力学的结果相同(能量曲线平行),显然不需要、也不可能进行“控制”;只有动力学和热力学两种因素的结果相反时(能量曲线交叉),对反应的动力学控制和热力学控制才有实际意义。

能量曲线平行 能量曲线交叉
例如:
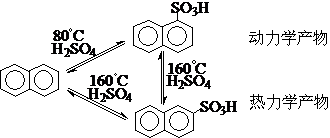

过渡态理论从能量角度讨论反应过程,在能量变化上,化学反应类似于“爬山运动”-能量变化曲线:
1、能量曲线剖面图:以能量E为纵轴,以反应进程为横轴,描述反应体系能量变化的曲线-能量曲线刨面图。

共价键的断裂和生成同步 共价键的断裂和生成不同步
能量上有利 能量上不利
有立体专一性需要 无立体专一性需要
主要认识有机化学反应的特殊性及其相关的理论
有机化学反应的特殊性:反应速度慢、副反应和副产物多-原因是:
分子体积大,活性中心多。
3、构象异构:
产生构象的原因:单键的“自由”转动
(1)碳链的构象:
乙烷:
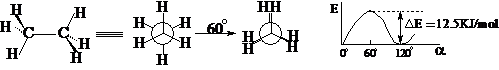
交叉式 重迭式
交叉式:前后H原子之间距离较大,斥力较小,内能较低;处于能量曲线的谷值位置(极小值),体系向前或向后变化,都导致内能升高,违背最低能量原理,所以体系可以在这一点停留,其寿命-半衰期 T1/2 > 0.
重迭式:前后H原子之间距离较小,斥力较大,内能较高;处于能量曲线的峰值位置(极大值),体系向前或向后变化,都导致内能升降低,符合最低能量原理,所以体系不可能在这一点停留,其寿命-半衰期 T1/2 = 0。
在交叉式和重迭式之间存在无数中其它构象,它们的能量处于交叉式和重迭式之间。所以在通常条件下,乙烷实际上是无数不同构象的热力学平衡体系,其中内能较低的交叉式为主,各种构象不可分离;只有在缺乏能量供给的情况下(如:-170℃),乙烷才能完全以交叉式存在。
丁烷:
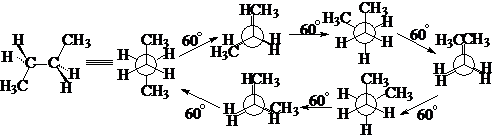
反交叉式 部分重迭式 顺交叉式(旁式) 全重迭式
内能最低 内能较高 内能较低 内能最高
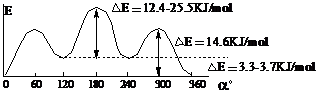
在室温下,丁烷的各种构象构成平衡体系,其中反交叉式为主,顺交叉为次,其它构象只具有理论意义。
锯齿型碳链:相当于反交叉式,内能最低,所以主要碳链以锯齿型存在。
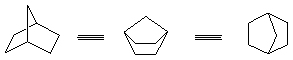 弓型碳链:相对于全重迭式,内能最高,所以游离的碳链不可能以弓型存在;刚性环中,碳链可以被迫以弓型存在,导致内能较高,如:二环[2.2.1]庚烷体系。
弓型碳链:相对于全重迭式,内能最高,所以游离的碳链不可能以弓型存在;刚性环中,碳链可以被迫以弓型存在,导致内能较高,如:二环[2.2.1]庚烷体系。
前视图 侧视图 俯视图
(2)环的构象
三元环-弯键学说
由于几何原因,三元环只能是平面环。
现代价键理论认为,三元环的每一个碳原子都是正常的SP3杂化,只是重迭方向与键轴不重合,重迭之后形成的电子云中心在键轴之外,形成弯型s键-香蕉键。所以三元环的单键具有两重性:由SP3杂化轨道重迭形成-s键的特点;电子云中心在轴外-p键的特点;因此三元环具有烷烃和烯烃两重性:象烷烃一样不能被KMnO4氧化剂氧化;象烯烃一样,易与X2、HX等亲电试剂,发生开环亲电加成反应。


开环位置:最多与最少之间;加成方向:服从马氏规则
四元环的构象:
现代电子衍射技术显示,四元环是非平面动态环,形如蝴蝶,两翼不停摆动,所以称为蝶式环。但在处理立体化学问题是,可以把四元环当成平面环。
五元环的构象:
现代电子衍射技术显示,非取代的环戊烷以信封式和扭船式两种构象存在:
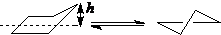
h=0.05nm
信封式 扭船式
一取代的环戊烷,主要以平伏取代的信封式构象存在:

六元环的构象:

椅式 半椅式 扭船式 船式
内能:椅式<扭船式<半椅式<船式
在一般情况下,环己烷环主要以椅式存在;有时可能以扭船式存在,如环己烯和苯并环己烷等(为什么?)。当然在刚性环中,六元环以船式存在。
竖直键a和平伏键e:当环己烷环从一个椅式变成另一个椅式时,e和a键发生互变。
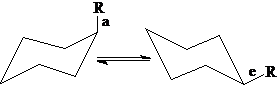
取代环己烷的构象及其内能
一取代环己烷:1,3-斥力导致a键取代内能较高,e键取代无1,3-斥力,内能较低。
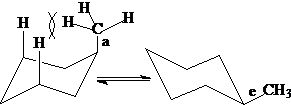
多取代环己烷:e键取代较多的构象内能较低
注意:构象互变时,构型不变,取代基之间的顺反关系不变。
葡萄糖的开链式(Fischer投影式)与环状式(哈沃斯式)
葡萄糖的a-吡喃式和b-吡喃式的构象和内能

a-型,5e+1a b-型,6e
葡萄糖溶液的平衡体系、变旋现象和化学反应

平衡体系的[α]D=+52°。氧化、还原、亲核加成(HCN、成脎反应等)是开链式的反应;酯化、苷化反应是环状式的反应。如:

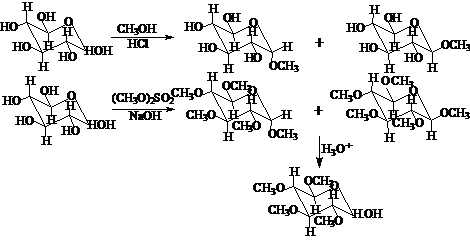
讨论分子和活性中间体的内能和反应活性的有关理论
2、构型异构:
非键合原子或原子团之间的空间关系,在平面内出现的为顺反异构(几何异构),涉及三维空间的为对映异构(立体异构、旋光异构、光学异构)。
(1) 顺反异构:凡是含有C=C、C=N(肟,腙等)的分子都可能有顺反异构现象,但脂环的化合物的顺反异构应当属于对映异构(SP3)。烯烃存在顺反异构的条件是:

a¹b同时c¹d
顺反异构体的数目=2n,n为链内双键的数目。
顺反异构体的命名:
顺、反:判断双键顺或反的标准是:相同的原子或原子团;
Z、E:判断双键Z或E的标准是:原子或原子团的大小次序(次序规则);
由于标准不同,所以“顺反”与“Z/E”没有直接关系,如:
虽然反-2-丁烯和E-2-丁烯相同,但是反-2-氯-2-丁烯和Z-氯-2-丁烯是同一个化合物。而有些顺反异构体只能用Z、E,不能用顺反命名,如肟类化合物和具有以下特征的烯烃:
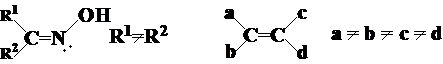
(2)对映异构(立体异构、旋光异构或光学异构)
对映异构的几何基础:
按几何特征,可以把所有的宏观物体分为两类:
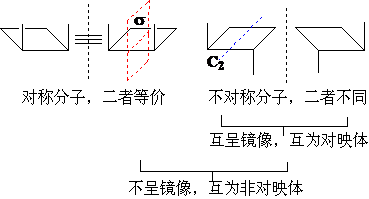
对称物体:与其镜像可以重合,属于同种物体; 如人:张三的镜像是张三。
对称物体:与其镜像不能重合,属于不同物体; 如手;左手的镜像是右手,二者不能重合,可以区别。
如果把这种理念引申到微观领域,那么我们很容易理解:
对称分子与其镜像可以重合,是同一种分子;如:甲烷的镜像仍然是甲烷,所以甲烷没有立体异构体。
不对称分子与其镜像不能重合,是不同的分子;如:D-乳酸的镜像是L-乳酸,二重不能重合,属于不同的分子,二者的关系属于对映异构或立体异构体。
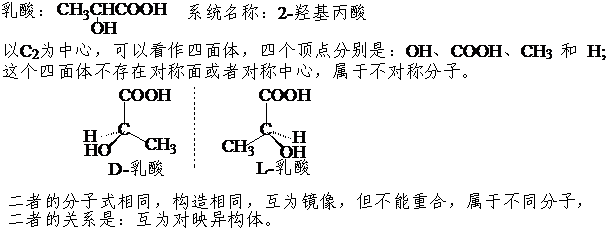
对称分子和不对称分子的几何判据
对称元素:对称面s、对称中心i 、对称轴Cn。
凡是具有s和/或i 的分子属于对称分子;
凡是没有s和 i 的分子属于不对称分子;
分子是否对称,与Cn无关!
例如:1,2-二甲基环丁烷,有三个异构体
不对称碳原子-手性碳原子的条件:
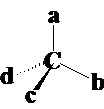
条件① SP3杂化;②a≠b≠c≠d
含有n个手性碳的化合物,一般有 2n 个立体异构体。如:葡萄糖的构造式为2,3,4,5,6-五羟基己醛,含有4个手性C(在结构式中以*标出),有24 = 16种立体异构体。
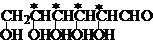
手性化合物的构型式:Fischer投影式
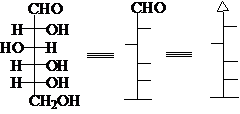
自然存在的D(+)-葡萄糖的Fischer投影式
Fischer投影式的原始规定:竖直向后,水平向前。
习 惯:碳链竖起来,C1在上。
异构体数目减少的因素:①相同的手性碳;②环的刚性
酒石酸:2个相同的手性C,3个异构体
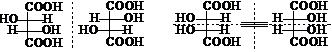
二环[2.2.1]-2-庚酮 :2个桥头手性C , 2个异构体
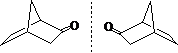
手性碳的构型:R、S
如果a > b > c > d(次序规则),那么对着最小的d 观查分子,从最大的a,经过中等的b,到小的c,顺时针为R,逆时针为S。
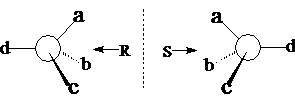
Fischer投影式常用来表示静态分子的构型,而Newman投影式常用来表示反应过程中的立体化学,因此Fischer投影式和Newman投影式的相互转化是立体化学的一项基本功,应当熟练掌握。
立体异构体的命名:系统名称:(手性碳的位次和构型)-构造名称

(2R,3S,4R,5R)-2,3,4,5,6-五羟基己醛
在投影式上判断R、S要注意最小原子团的方向:
最小原子团在竖直方向时,纸面构型与实际相同!
最小原子团在水平方向时,纸面构型与实际相反!
投影式不能随便放倒 ,放到导致前后关系发生变化不再代表原来的异构体。如:

2R,3S,4R,5R
2S,3R,4S,5S
相对构型(D、L)
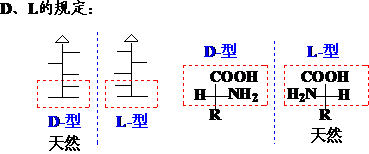
使用D、L表示构型时,习惯用化合物的俗名或习惯名称:

立体异构体性质的差别及其应用
对映体:在对称的条件下(环境、溶剂、试剂等)性质相同;在不对称的条件下(物理、化学)性质不同。
非对映体:在任何条件下性质都不相同。
内消旋体:对称分子,纯净化合物,不能拆分。
外消旋体:对映体的等量混合物,可以拆分。
区别或者分离对映体的基本原则:先要把它们变成非对映体,然后再分离或者检测
外消旋体的拆分原理:
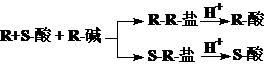
对映体的定性和定量分析原理:
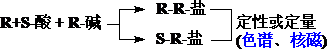
湖北省互联网违法和不良信息举报平台 | 网上有害信息举报专区 | 电信诈骗举报专区 | 涉历史虚无主义有害信息举报专区 | 涉企侵权举报专区
违法和不良信息举报电话:027-86699610 举报邮箱:58377363@163.com