[氢]元素符号H,原子序数1,原子量1.0079,核外电子排布式为1s1,氢的电离能为1306.47千焦/摩尔,电负性为2.1。氢在元素周期表中位于第一周期第Ⅰ主族或第Ⅶ主族。氢在自然界中有三种同位素,氕、氘、氚,其中氢-1(1H,氕)相对丰度为99.985%,氢-2(2H,氘也叫重氢)相对丰度为0.016%,这两种氢是在自然界中稳定的同位素。从核反应中还找到质量数为3的同位素氢-3(3H,氚,也叫起重氢),它在自然界中含量极微。氢在自然界中主要以化合物存在,其中以水和碳氢化合物为主,在大气中含量极微(体积百分比为5×10-5%),在地壳里的丰度为0.76%。根据光谱分析在太阳和某些星球的大气中含有大量氢气。1776年,英国化学家卡文迪许用金属跟酸作用时发现了氢。氢原子是结构最简单的原子。氢是元素中最轻的元素。氢气的分子式为H2,氢气在通常状况下为无色、无气味的气体,氢气的密度为0.0899克/升,液氢的密度为0.070克/厘米3,熔点-259.14℃,沸点-252.4℃,氢气是最轻的气体(密度最小),微溶于水(0℃时,每体积水溶解0.0214体积氢气)。氢气有最大的扩散速度,容易通过各种细小的空隙,如钢板。高扩散速度使氢气有高导热性。用氢气冷却物体比用空气冷却约快6倍。氢分子通过电弧或高温,都能离解成氢原子,氢原子在金属表面很快又结合成氢分子,同时放出大量的热,形成氢原子火焰,温度可高达3500℃。
氢气有可燃性,它在空气中燃烧生成水并放出大量的热,当空气中含4.1-75%(体积百分比)氢气时,点火则发生爆炸。氢气跟氧气按一定比例混和时即得爆鸣气,因此,点燃氢气前必须检验氢气的纯度,保证安全。氢气可以跟氟、氯、溴、碘、硫、氮等很多非金属化合生成气态氢化物,其中氢为+1价。氢气还能跟一些碱金属、碱土金属化合生成固态金属氢化物如:LiH、NaH、KH、CaH2、BaH2等,在这些化合物中氢为-1价。这些氢化物属于离子型化合物。氢气有还原性,在一定温度下,氢气可以从某些金属氧化物或非金属氧化物中夺取氧。用氢气可使氧化钨还原为金属钨。在一定温度下,也可从某些金属氯化物或非金属氯化物中夺取氯,使金属或非金属还原出来。氢气还可跟不饱和烃及其衍生物发生加成反应。钯和某些合金如LaNi5等能大量“吸收”氢气,在一定条件下氢气又可以被释放出来,因此钯和这些合金可用做氢气的储运材料。
氢气或氢氦混和气体可用来充填气球,氢气可用来制燃料电池,液氢和液氧可用做火箭的高能燃料,氢原子焰可用来焊接金属。利用氢气的还原性可用来冶炼金属或制纯硅。氢气是重要的化工原料,可用于合成氨、合成盐酸以及有机合成等。
氢气可用下列几种方法制取:
⑴电解氢氧化钠水溶液,制得的氢气纯度较高(99.9%);
⑵甲烷转化法,甲烷和水蒸气在高温和镍、钴催化作用下转化成氢气和一氧化碳;
⑶水煤气法,用焦炭在高温下跟水蒸气反应;
⑷离子型金属氢化物如氢化钙跟水反应;
⑸用过渡金属络合物做催化剂,利用太阳能分解水制取氢气是发展氢能源的方向;
⑹在实验室中常用锌跟稀硫酸或稀盐酸作用制取少量氢气。
[重氢]符号21H(或D),又名氘(音刀),是氢的同位素,质量数为2,原子核中有一质子和一中子。常温下为气体,分子式为D2,分子量为4。沸点-249.7℃,熔点-254.6℃,都高于氢气。
重氢(氘)的化学性质跟氢(氕)相似,但活动性较氢差,跟氧化合生成重水(D2O),与氮化合生成重氨(ND3)。重氢主要存在于重水中,氢气中含重氢约0.02%。重氢通常通过电解重水,或者通过重水跟锌、铁、钙等反应制得。分馏液态氢,氕先蒸馏出来,剩下氘。人工加速的重氢原子核(即由一个质子和一个中子组成的原子核-氘核)能参与许多核反应。重氢(氘)和超重氢(氚)在极高温度(如1亿K)可发生核聚变反应生成氦,同时释放出巨大能量。核聚变反应也叫热核反应。重氢是制造氢弹的核材料,也是可控热核反应的核燃料。
[超重氢]符号31H或T,原子核由一个质子和两个中子组成,质量数为3,是氢的唯一的放射性同位素。在通常状况下T2是气体,熔点-252.54℃,沸点-248.12℃。化学性质和氢(氕)相似,因为氚比氕重,所以氚的许多反应比氕要慢得多。氚能发射β粒子衰变成质量数为3的氦核,在衰变过程中不发射γ射线,半衰期为12.26年。氚和氘可发生热核反应,放出巨大能量。氚有放射性可用做研究化学反应过程和生物体中物质变化过程时的示踪原子。用高能氘核轰击氘化合物可得到氚。锂的同位素63Li吸收慢中子可产生氚。
[氕]氢的同位素,符号11H。原子核由一个质子组成,详见氢条。
[氘]氢的同位素,详见重氢条。
[氚]氢的同位素,详见超重氢条。
[水]分子式为H2O,水在地球表面上分布最广,约占地球表面的3/4,大气中含有水蒸气,土壤、岩石、动植物体中也含有大量的水,动物体中的水约占70%,新鲜植物体中的水分约占80-90%,地球上水的含量约有2×1018吨。
18世纪以前,人们认为水是一种单质,1781年英国化学家卡文迪许首先发现氢气在空气中燃烧生成的唯一产物是水,证明水是由氢、氧两种元素组成的化合物。几年以后,拉瓦锡测定了水的质量组成。近代结构理论证明,水分子呈V形结构。用X射线测定水的晶体(冰)结构,证明两个O-H键间形成104.5°的键角,由于水分子是不对称结构,故知水是极性分子。实验还证明,水分子通过氢键发生缔合,形成较复杂的分子集团,水沸腾时水蒸气中含有3.5%的双分子水(H2O)2,液态水中含有较复杂的(H2O)n分子(n可以是2、3、4…),这些较复杂的缔合水分子的化学性质和水(H2O)相同。
纯水是无色、无嗅、无味的液体,深层的天然水呈蓝绿色。水的热容为4.1868焦尔/克·度(1卡/克·度),在所有液态和固态物质中,水的热容最大。水在4℃时密度为1克/厘米3。在101325帕(1大气压)时,水的沸点为100℃,水的气化热为2.257千焦/克,凝固点是0℃。水是极弱的电解质,可发生自偶离解:
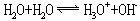
常温下,水的电离常数为1.8×10-16,水的离子积Kw=1×10-14。
水是较稳定的化合物,在1000℃以上才开始分解。在硫酸或氢氧化钠等存在条件下,电解水可生成氢气和氧气。许多活动性强的金属如K、Na、Ba、Ca等在常温下可跟水反应生成碱和氢气,Mg、Al在加热至水沸腾时跟水反应生成碱和氢气。水可跟许多较活动金属如锌、铁等在高温下反应生成金属氧化物和氢气。水可跟某些非金属反应,如:
2F2+2H2O=4HF+O2↑
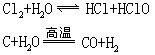
水可以跟许多碱性氧化物如K2O、Na2O、CaO、BaO、MgO等化合生成碱,水可以和多种酸性氧化物SO3、SO2、CO2、P2O5等化合生成相应的酸。水还能跟某些盐或酯、糖等有机物发生水解反应。
水是一种广泛应用的溶剂,极性物质(如HCl、NaCl等)容易在水中溶解。
[重水]化学式D2O,是氢的同位素重氢(氘)和氧的化合物。式量20.31。普通水中约含重水0.015%。重水是无色、无嗅的液体,密度是1.105克/厘米3,熔点3.82℃,沸点101.42℃。重水在原子核反应堆中用做中子的减速剂,也可用做制取重氢(氘)的原料。大量重水是以天然水为原料,通过电解、蒸馏、与硫化氢双重温度交换再电解等方法制取并富集的。
[双氧水]见过氧化氢条。
[过氧化氢]化学式H2O2,式量34。俗名双氧水。纯者为淡蓝色粘稠液体,密度1.465克/厘米3,熔点-0.41℃,沸点150.2℃。商品一般为30%的水溶液,医疗用3%的稀溶液做消毒剂,洗涤伤口。在过氧化氢分子中有一个过氧原子团-O-O-。每个氧原子各连结一个氢原子。常温时纯过氧化氢相当稳定,当加热到153℃以上时,纯过氧化氢即发生爆炸,分解生成水和氧气。过氧化氢及其水溶液经阳光照射或混有重金属离子(如Mn2+、Cu2+、Cr3+、Fe2+)时都能加速分解。过氧化氢中氧的氧化数是-1,它有向-2或0价态转化的两种可能性,因此,它既有氧化性又有还原性。过氧化氢在酸性介质或碱性介质中都是强氧化剂,它主要用做氧化剂;但若遇到更强的氧化剂时(如KMnO4),在酸性或碱性介质中,它即用做还原剂。纯过氧化氢可用作某种火箭的氧化剂和推进剂,纺织工业中用作漂白剂。可用硫酸或酸式硫酸盐电解氧化生成过硫酸或过硫酸盐,再经水解后制得;工业上还可以用乙基蒽醌法来制备过氧化氢。
[络合物]又称配位化合物。凡是由两个或两个以上含有孤对电子(或π键)的分子或离子作配位体,与具有空的价电子轨道的中心原子或离子结合而成的结构单元称络合单元,带有电荷的络合单元称络离子。电中性的络合单元或络离子与相反电荷的离子组成的化合物都称为络合物。习惯上有时也把络离子称为络合物。随着络合化学的不断发展,络合物的范围也不断扩大,把NH4+、SO42-、MnO4-等也列入络合物的范围,这可称作广义的络合物。一般情况下,络合物可分为以下几类:
⑴单核络合物,在1个中心离子(或原子)周围有规律地分布着一定数量的配位体,如硫酸四氨合铜[Cu(NH3)4]SO4、六氰合铁(Ⅱ)酸钾K4[Fe(CN)6]、四羧基镍Ni(CO)4等,这种络合物一般无环状结构。
⑵螯合物(又称内络合物),由中心离子(或原子)和多齿配位体络合形成具有环状结构的络合物,如二氨基乙酸合铜:
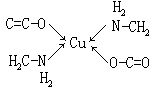
螯合物中一般以五元环或六元环为稳定。
⑶其它特殊络合物,主要有:多核络合物(含两个或两个以上的中心离子或原子),多酸型络合物,分子氮络合物,π-酸配位体络合物,π-络合物等。
[配位化合物]见络合物条。
[中心离子]在络合单元中,金属离子位于络离子的几何中心,称中心离子(有的络合单元中也可以是金属原子)。如[Cu(NH3)4]2+络离子中的Cu2+离子,[Fe(CN)6]4-络离子中的Fe2+离子,Ni(CO)4中的Ni原子等。价键理论认为,中心离子(或原子)与配位体以配位键形成络合单元时,中心离子(或原子)提供空轨道,是电子对的接受体。
[配位体]跟具有空的价电子轨道的中心离子或原子相结合的离子或分子。一般配位体是含有孤对电子的离子或分子,如Cl-、CN-、NH3、H2O等;如果一个配位体含有两个或两个以上的能提供孤对电子的原子,这种配位体称作多齿配位体或多基配位体,如乙二胺:
H2N-CH2-CH2-NH2,三乙烯四胺:H2N-C2H4-NH-C2H4-NH-C2H4-NH2
等。此外,有些含有π键的烯烃、炔烃和芳香烃分子,也可作为配位体,称π键配位体,它们是以π键电子与金属离子络合的。
[络离子]见络合物条。
[内界]在络合物中,中心离子和配位体组成络合物的内界,通常写在化学式的[ ]内加以标示,如:
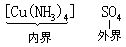
[外界]络合物内界以外的组成部分称外界。如[Cu(NH3)4]SO4中的SO42-离子。外界离子可以是阳离子,也可以是阴离子,但所带电荷跟内界络离子相反。在络合物中外界离子与内界络离子电荷的代数和为零。
[配位数]在络合单元中,一个中心离子(或原子)所能结合的配位体的配位原子的总数,就是中心离子(或原子)的配位数。如[Fe(CN)6]4-中,Fe2+是中心离子,其配位数为4,二氨基乙酸合铜(见络合物)中Cu2+是中心离子,它虽然与两个二氨基乙酸离子络合,但是直接同它络合的共有4个原子(2个N原子,2个O原子),因此Cu2+的配位数也是4。
[配位原子]配位体中具有孤对电子并与中心离子(或原子)直接相连的原子。
[单齿配位体]又称单基配位体,是仅以一个配键(即孤电子对)与中心离子或原子结合的配位体。如[Ag(NH3)2]+中的NH3分子,[Hgl4]2-中的I-离子,[Cu(H2O)4]2+中的H2O分子等。
[单基配位体]见单齿配位体条。
[多齿配位体]又称多基配位体,若一个配位体含有两个或两个以上的能提供孤电子对的原子,这种配位体就叫多齿配位体。如乙二胺H2CH2-CH2-H2,乙二胺四乙酸酸根离子(EDTA):
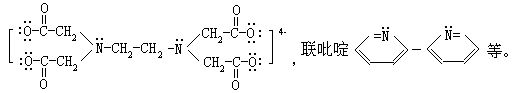
[多基配位体]见多齿配位体条。
[螯合物]见络合物条。
[螯环]螯合物中所形成的环状结构。一般以五元环和六元环为稳定。
[螯合剂]能够提供多齿配位体和中心离子形成螯合物的物质。
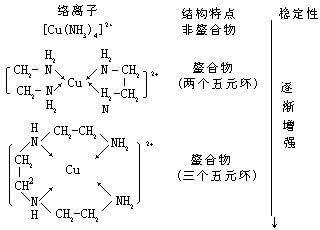
[螯合效应]对同一种原子,若形成螯合物比单基配位体形成的络合物(非螯合物)要更加稳定,这种效应称作螯合效应。螯合物一般以五元环、六元环为最稳定,且一个络合剂与中心离子所形成的螯环的数目越多就越稳定。以铜离子Cu2+和氨分子及胺类形成的络合物为例:
[内轨型络合物]价键理论认为中心离子(或原子)和配位体以配位键结合,中心离子(或原子)则以杂化轨道参与形成配位键。若中心离子(或原子)以(n-1)d、ns、np轨道组成杂化轨道与配位体的孤对电子成键而形成的络合物叫内轨型络合物。如[Fe(CN)6]4-离子中Fe2+以d2sp3杂化轨道与CN-成键;[Ni(CN)4]2-离子中Ni2+以dsp2杂化轨道与CN-成键。内轨型络合物的特点是:中心离子(或原子)的电子层结构发生了变化,没有或很少有末成对电子,因轨道能量较低,所以一般内轨型络离子的稳定性较强。
[外轨型络合物]若中心离子(或原子)以ns、np、nd轨道组成杂化轨道与配位体的孤对电子成键而形成的络合物叫外轨型络合物。如[FeF6]3-离子中Fe3+以sp3d2杂化轨道与F-成键;[Ni(H2O)6]2+离子中Ni2+以sp3d2杂化轨道与H2O成键。有的资料把中心离子以ns、np轨道组成的杂化轨道和配位体成键形成的络合物也称作外轨型络合物,如[Zn(NH3)4]2+离子中,Zn2+以sp3杂化轨道与NH3成键。外轨型络合物的特点是:中心离子(或原子)电子层结构无变化,未成对电子数较多,因轨道能量较高,所以一般外轨型络合物的稳定性较差。
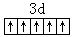
[低自旋络合物]含有较少的未成对电子的络合物,一般是内轨型络合物。这种络合物的中心离子的未成对电子数目,一般比络合前有所减少,如[Fe(CN)6]3-中,Fe3+离子在未络合前3d亚层有5个未成对电子:
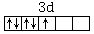
而在此络离子中Fe3+离子的3d亚层上只有1个未成对电子:
[高自旋络合物]含有较多的未成对电子的络合物,一般是外轨型络合物。这种络合物的中心离子的未成对电子数目,在络合前后一般保持不变。如[FeF6]3-络离子中Fe3+离子仍含有5个不成对电子。
[络合平衡]溶液中存在的络离子(或络合分子)的生成与离解之间的平衡状态。例如:
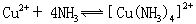
当络离子的生成与离解达到平衡状态时,虽然两个相反过程还在进行,但它们的浓度不再改变。
[稳定常数]络合平衡的平衡常数。通常指络合物的累积稳定常数,用K稳表示。例如:
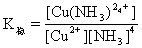
对具有相同配位体数目的同类型络合物来说,K稳值愈大,络合物愈稳定。
[逐级稳定常数]络合物的生成一般是分步进行的。对应于这些平衡也有一系列的稳定常数,每一步的稳定常数就是逐级稳定常数。例如,[Cu(NH3)4]2+的生成(或解离)分四步:
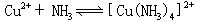
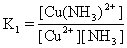
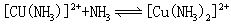
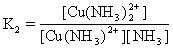
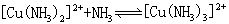
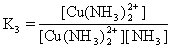
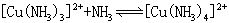
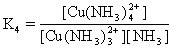
K1、K2、K3、K4就是[Cu(NH3)4]2+的逐级稳定常数,逐级稳定常数的乘积就是累积稳定常数。
K稳=K1·K2·K3·K4
lgK稳=lgK1+lgK2+lgK3+lgK4
[不稳定常数]络合物的不稳定常数用K不稳表示,与稳定常数成倒数关系:
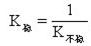
以络合物的离解的方向为正方向,如:
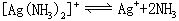
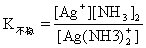
对具有相同数目配位体的同类型络合物来说,K不稳愈大,络合物愈易离解,即愈不稳定。
[络酸]外界离子是氢离子,在溶液中能电离产生氢离子而显酸性的络合物。如氯铂酸即六氯合铂(Ⅳ)酸H2[PtCl6]:
H2[PtCl6]→2H++[PtCl6]2-
[络碱]外界离子是氢氧离子OH-,在溶液中能电离产生OH-而显碱性的络合物。如氢氧化四氨合铜(Ⅱ)[Cu(NH3)4](OH)2:
[Cu(NH3)4](OH)2→[Cu(NH3)4]2++2OH-
[络盐]又称错盐,指含有络离子的盐类。例如K4[Fe(CN)6]、[Ag(NH3)2]Cl、[Cu(NH3)4]SO4等。络盐中的络离子,在溶液中较稳定,很难离解,这是络盐和复盐的重要区别。
[错盐]见络盐条。
[维尔钠配位理论]1893年由瑞士化学家维尔纳(Wer-ner)提出。其要点是:
⑴一些金属的化合价除主价外,还可以有副价。例如在CoCl3·4NH3中,钴的主价为3,副价为4,即三个氯离子满足了钴的主价,钴与氨分子的结合使用了副价。
⑵络合物分为“内界”和“外界”,内界由中心离子与周围的配位体紧密结合,而外界与内界较易解离。例如CoCl3·4NH3可写成[Co(NH3)4Cl2]Cl,内界是[Co(NH3)4Cl2]+,外界是Cl-。
⑶副价也指向空间的确定方向。维尔纳的配位理论解释了大量的实验事实,但对“副价”的本质未能给以明确的解释。
[络合物的价键理论]络合物的化学键理论之一。其要点如下:
⑴中心离子(或原子)提供空轨道,配位体提供孤对电子,以配位键结合。
⑵中心离子(或原子)参与成键的空轨道都是杂化轨道,具有一定的饱和性和方向性。
⑶中心离子(或原子)提供杂化轨道接受配位体的孤对电子形成配位键时,由于采用的能级轨道不同,形成的络合物分为外轨型和内轨型。
若中心离子(或原子)以ns、np、nd轨道组成杂化轨道和配位原子形成配位键时,就叫外轨型络合物,如[FeF6]3-;中心离子(或原子)以(n-1)d、ns、np轨道组成杂化轨道和配位原子形成配位键时,则叫内轨型络合物,如[Fe(CN)6]4-。
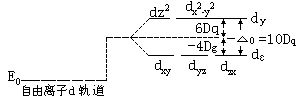
[络合物的晶体场理论]络合物的化学键理论之一。是1923-1935年由培特(H.Bethe)和冯弗莱克(J.H.Van Vleck)提出了晶体场理论(CFT),本世纪50年代晶体场理论又发展成配位场理论(LFT)。晶体场理论的基本观点是:认为中心离子和配位体之间的相互作用是静电作用。它的要点如下:
⑴中心离子原来简并的d轨道在配位体电场的作用下,发生了能级分裂,有的能量升高,有的能量降低。分裂后,最高能量d轨道和最低能量d轨道之间的能量差叫分裂能。中心离子的d轨道能量在正八面体场中的分裂如下图所示:
中心离子的d轨道能量在正四面体场中的分裂如下图所示:
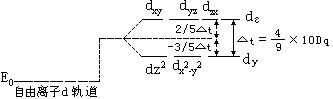
⑵分裂能Δ值的大小,主要受配位体的电场、中心离子的电荷及它属于第几过渡系等因素的影响。
⑶使本来是自旋平行分占两个轨道的电子挤到同一轨道上去必会使能量升高,这增高的能量称为成对能,用Ep表示。在弱配位场中Δ<Ep,d电子尽可能占据较多的轨道且自旋平行,形成高自旋络合物;在强配位场中Δ>Ep,d电子尽可能占据能量较低的轨道形成低自旋络合物。
[晶体场稳定化能]在晶体场理论中将d电子从未分裂的d轨道进入分裂的d轨道所产生的总能量的下降值,称为晶体场稳定化能(CFSE)。总能量下降愈多,即CFSE愈大(负值绝对值愈大),络合物就愈稳定。
[络合物的分子轨道理论]络合物的化学键理论之一。化学键的分子轨道理论的基本观点,在这里都是完全适用的。分析中心离子(原子)和配位体组成分子轨道,通常按下列步骤进行:
⑴找出中心离子(原子)和配位体的价电子轨道,按所组成的分子轨道是σ轨道还是π轨道分组,分别称为σ轨道和π轨道。
⑵将配位体中的σ轨道和π轨道分别重新组合成若干新轨道,这些新轨道称为群轨道,使得这些群轨道的对称性分别与中心离子(原子)的各原子轨道相匹配。
⑶将对称性相同的中心离子(原子)的原子轨道和配位体的群轨道组合成分子轨道。络合物的分子轨道理论可以得到和晶体场理论一致的结果,同时又能解释光谱化学系列、有机烯络合物的形成、羰基络合物的稳定性等方面的问题。
[分散体系]即分散系。一种或几种物质分散在另一种物质里所组成的体系。分散体系中被分散的物质称为分散质或分散相;分散其它物质的物质称为分散剂或分散介质。按分散相微粒的大小,常把分散体系分为三种:真溶液、胶体、粗分散系。分散系的另一种分类法,是按聚集状态区分,可分为九类:
|
|
⑴ |
⑵ |
⑶ |
⑷ |
⑸ |
⑹ |
⑺ |
⑻ |
⑼ |
|
分散介质 |
气 |
气 |
气 |
液 |
液 |
液 |
固 |
固 |
固 |
|
分散相 |
气 |
液 |
固 |
气 |
液 |
固 |
气 |
液 |
固 |
|
实例 |
空气 |
云雾 |
烟尘 |
各种泡沫 |
酒精水溶液 |
糖水 |
泡沫塑料 |
硅凝胶 |
合金 |
[分散剂]见分散体系条。
[分散介质]即分散剂。
[分散质]见分散体系条。
[分散相]即分散质。
[悬浊液]粗分散系的一种,是不溶性固体分散相以微小颗粒分散在液体物质中形成的分散体系。分散相微粒的线性大小在10-7-10-3米之间。悬浊液是不均一、不稳定的混和物。久置,固体分散相会沉淀下来,如泥水是悬浊液。
[乳浊液]粗分散系的一种,是互不相溶(或不完全相溶)的液体组成,液体分散相以微小珠滴分散在另一种液体物质中形成的分散体系。分散相的微粒的线性大小在10-7-10-3米之间。乳浊液通常分为两种类型:水包油型,以O/W表示;油包水型,以W/O表示。乳浊液是不均一、不稳定的混和物,久置会分层,如果加入乳化剂可增加其稳定性。
[溶液]物质以分子或离子的状态,均匀地分布在另一种物质中形成的分散体系。分散相的微粒是分子或离子,其线性大小在10-9-10-10米。溶液是均一、稳定的混和物,只要外界条件不发生变化(温度不变、溶剂没有蒸发),长久放置,溶质也不会析出。溶液又称分子溶液或真溶液。溶液有液态溶液(如蔗糖水、食盐水、碘酒等),固态溶液(如合金),气态溶液(如气体混和物)三种。
[溶质]溶液中被溶解的物质叫溶质。溶质可以是气体、液体、固体。通常当气体或固体溶解在液体中时,气体或固体是溶质。如液体溶于液体时,以量少者为溶质。
[溶剂]溶液中使其它物质溶解的物质叫溶剂。通常当气体或固体溶解在液体中时,液体为溶剂。如液体溶于液体,以量多者为溶剂。根据溶剂分子的特征,溶剂有极性溶剂(如水)和非极性溶剂(如汽油、苯、四氯化碳等)之分。
[溶解]溶质分散在溶剂中形成溶液的过程。物质溶解在水里,通常发生两种过程:一种是溶质分子(或离子)的扩散过程,这种过程吸收热量,是物理过程;另一种是溶质分子(或离子)和水分子作用,形成水合分子(或水合离子)的过程,这种过程放出热量,是化学过程。因此,在溶解过程中往往伴有热量、体积及颜色的改变。例如,硝酸铵溶于水吸热多于放热,溶液的温度降低;硫酸溶于水放热多于吸热,溶液的温度升高;白色的无水硫酸铜溶于水形成蓝色溶液;酒精和水混和溶液总体积减小。
[结晶]晶体从溶液(液态或气态)中析出的过程。结晶也是常用的提取或提纯物质的一种方法。结晶的方法主要有减少溶剂和降低温度两种。前者用于温度对溶解度影响不大的物质,如用蒸发的方法从海水中提取食盐;后者用于温度对溶解度影响较大的物质,如从氯化钠和硝酸钾的混和溶液中提取硝酸钾。
[母液]在化学结晶或沉淀的过程中,分离出结晶或沉淀后,残余的饱和溶液。
[晶种]在结晶过程中加入的预先制成的晶体微粒。
[相似相溶规律]当物质溶解在溶剂中时,极性分子易溶于极性溶剂,非极性分子易溶于非极性溶剂。例如碘分子I2是非极性分子,碘易溶于四氯化碳,因为CCl4分子也是非极性分子,碘在水中的溶解度就很小,因为H2O分子是极性分子。而氨在水中的溶解度就很大,因为NH3分子和H2O分子都是极性分子。相似相溶规律是从大量事实总结出来的经验规律,不能说明所有物质的溶解情况。这一规律可以由分子间作用力和化学热力学的原理加以说明。
[溶解平衡]一般指在一定条件(温度、压力)下,固体溶质在液体溶剂中溶解的速度与溶液中溶质结晶析出的速度相等时的状态。溶解平衡也是一种动态平衡。
[饱和溶液]在一定温度和压力下,达到溶解平衡时的溶液。饱和溶液中新加入的溶质不能继续溶解,已溶解的溶质也不会析出。
[不饱和溶液]在一定温度和压力下,小于该条件下饱和溶液浓度的溶液。在不饱和溶液中加入新的溶质能继续溶解。
[过饱和溶液]在一定温度和压力下,大于该条件下饱和溶液浓度的溶液。过饱和溶液是个不稳定体系。在过饱和溶液中加入溶质后,新加入的溶质不能溶解,已溶解的部分溶质也会析出。过饱和溶液被振荡或用玻璃棒摩擦器壁,溶液中过量的溶质也会结晶析出。通常,过饱和溶液是在较高温度下的饱和溶液中加过量溶质缓慢冷却后得到的,十水合硫酸钠、硼砂、硫代硫酸钠等易形成过饱和溶液。
[溶解度]在一定温度和压力下,物质在一定量溶剂中溶解的最高量(即达到饱和溶液状态时所溶解的量)。固体或液体物质的溶解度,通常以100克溶剂里所溶解溶质的最多克数来表示。气体物质的溶解度,则常用1体积(l毫升或1升)溶剂中所溶解溶质的最多体积数来表示。物质的溶解度除与溶质、溶剂的性质有关外,还与温度、压力有关。大多数固体或液体的溶解度随温度升高而增大,而气体溶解度随温度的升高而减小。压力一般对固体或液体的溶解度影响不大,而气体的溶解度却随压力增大而显著增大。
[溶液的浓度]一定量溶液或溶剂中所含溶质的量称为溶液的浓度。
[百分比浓度]即质量百分比浓度。用溶质的质量占全部溶液质量的百分比表示的溶液的浓度。溶液的百分比浓度用下式计算:

[体积比浓度]以液体试剂(原装)与溶剂体积之比来表示溶液浓度的方法。如将1体积原装的浓硫酸与5体积水混和得到的硫酸溶液浓度就是1:5,或记为(1:5)H2SO4。
[摩尔浓度]指体积摩尔浓度,以1升溶液里含有多少摩尔溶质来表示的溶液浓度。摩尔浓度的单位是摩尔/升或记成mol/L。摩尔浓度的计算公式是:
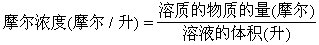
[质量摩尔浓度]以1千克溶剂中溶有多少摩尔溶质来表示的溶液的浓度,单位是摩尔/千克,或记成mol/kg。质量摩尔浓度的计算公式是:
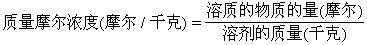
如在1千克溶剂中溶有0.2摩尔的溶质,此溶液的浓度为0.2mol/kg。
[质量体积浓度]以每升溶液中含有多少克溶质来表示溶液浓度的一种方法。如某溶液的浓度为5克/升,表示1升此溶液中含有5克溶质。
[当量浓度]以1升溶液中所含某溶质的克当量数表示的浓度。当量浓度在分析化学计算中有一定的作用,常以N表示。
[ppm浓度]溶液的浓度用溶质质量占溶液质量的百万分之比来表示的叫ppm浓度,即每千克溶液中含溶质的毫克数。这种浓度表示方法常用于极稀的溶液(如植物生长刺激素溶液)或自然环境、食物中有害物质含量的表示。
[ppb浓度]溶液的浓度用溶质质量占溶液质量的十亿分之比来表示的叫ppb浓度,即每千克溶液中含溶质的微克数。这种浓度表示方法用于极稀的溶液和某些含量极低的物质。
[等渗溶液]指渗透压相等的两种溶液。如0.9%的生理盐水跟人体里的血液渗透压相等,它们是等渗溶液。
[胶体]即胶体分散系,指分散相微粒线性大小在10-3-10-7米的分散系,这时分散相微粒比单个分子大得多,是由许多分子或离子组成的集合体,分散相与分散介质存在着相界面。所以说,胶体分散系是一个高分散度的多相体系。
[液溶胶]又称溶胶或胶体溶液,指分散介质是液体的胶体。如Fe(OH)3胶体、AgI胶体、墨水等。
[气溶胶]指分散介质是气体的胶体。如雾、云、烟等。
[固溶胶]指分散介质是固体的胶体。如烟水晶、有色玻璃等。
[胶体粒子]指胶体中的分散相微粒,又称胶粒。以液溶胶的胶体粒子为例分析,胶粒由胶核和吸附层组成,如在KI过量时形成的AgI胶体,AgI是胶核;胶核吸附了I-离子,I-离子叫电位离子,溶液中的反号离子K+有一部分进入吸附层,称反离子;还有一部分反离子疏散地分布在胶粒周围形成扩散层,见下图:
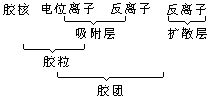
胶粒与扩散层一起组成胶团。若在制备AgI胶体时,AgNO3过量则胶粒、胶团的构成是:
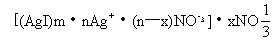
[高分子溶液]高分子化合物溶于适当的溶剂中可形成高分子溶液。高分子溶液具有双重性质,一方面由于这种分散相微粒大小与溶胶粒子相近,表现出溶胶的某些特性;另一方面高分子溶液是分子分散体系,又有某些真溶液的特点。高分子溶液和胶体溶液有许多不同之处:
⑴高分子溶液是单相体系,胶体是多相体系。
⑵高分子溶液分散相极易溶剂化,这是因为高分子化合物组成中,常含有大量亲水基团,如-OH、-COOH、-NH2等,而胶体微粒的溶剂化能力比高分子化合物弱得多。
⑶高分子溶液中分散相微粒一般不带电荷,胶体微粒则是带电的,高分子溶液的稳定性是它的高度溶剂化起了决定性作用。
⑷高分子化合物溶解的过程就是溶剂化过程,当用蒸发的方法除去溶剂后再加入溶剂仍能自动溶解,它的溶解过程是可逆的,而胶体中的胶粒一旦凝聚,一般很难或者不能用简单加入溶剂的方法使之复原。高分子溶液还有一项与真溶液和溶胶都不同的特性,就是有较大的粘度。
[丁达尔效应]当一束光线通过胶体溶液或高分子溶液时,因分散质微粒的散射作用,在入射光垂直方向,可看到一条明显的光柱,这种现象称丁达尔效应。由英国物理学家丁达尔(Tyndall)首先发现。
[布朗运动]1827年由英国生物学家布朗(Brown)发现,悬浮在水里的花粉小颗粒在不停地进行着不规则运动。这种现象叫布朗运动。溶胶中的胶体微粒也在进行布朗运动,这是由于周围分散剂的分子从各方面不均匀地撞击胶体微粒和胶体微粒本身的热运动造成的。布朗运动的速度取决于微粒大小、温度及介质的粘度,微粒越小、温度越高、介质粘度越小,布朗运动的速度越快。
[电泳]在外电场的作用下,胶体微粒向阳极(或阴极)作定向移动的现象。如三硫化二砷的胶体微粒带负电,会向阳极移动;氢氧化铁胶体微粒带正电会向阴极移动。电泳在工业生产、医疗诊断等方面都有重要的应用。
[渗析]又称透析。利用半透膜(膀胱膜、肠衣、羊皮纸、玻璃纸等)使溶胶和其中混有的离子或分子杂质分开的过程。将混有杂质的胶体放入有半透膜的装置内,并将此装置放在溶剂(如水)中,半透膜的细孔只能使离子或分子透过而不能使胶体微粒通过,这就可以使杂质离子和分子从溶胶中分离出来。应用渗析的方法可以精制某些胶体。
[半透膜]见渗析条。
[聚沉]又称胶体的凝聚。胶体具有一定的稳定性,在一定条件下,可以保存较长时间。但胶体是热力学不稳定体系,有自发聚结下沉的趋势,只要减弱或消除使胶体稳定的因素,胶体微粒就能聚集成较大颗粒而沉降。使胶体微粒聚集成较大的颗粒而沉降的过程叫聚沉。使胶体聚沉的方法主要有:加入电解质、加热、加入带相反电荷的胶体等。
[盐析]在大量电解质的作用下,使高分子化合物从溶液中析出的过程。例如往血清中加入(NH4)2SO4当浓度约为2.0摩尔/升时,其中球蛋白析出,当浓度为3-3.5摩尔/升时,血清蛋白析出。盐析可以看作是电解质对高分子溶液的聚沉作用,但盐析和溶胶聚沉有以下不同:
⑴盐析时电解质的用量很大。
⑵盐析结果生成的沉淀用溶剂加以冲淡仍可形成溶液。
⑶离子的价数对盐析能力的影响程度不象溶胶聚沉那样显著。
⑷盐析与离子电荷符号无关。
[胶凝]大分子溶液在一定条件下,粘度会逐渐变大,以致失去流动性而使整个体系变成一种弹性半固体的“冻”状,这个过程称胶凝。
[凝胶]胶凝过程中形成的“冻”状体系叫凝胶。
[高分子电解质]有些高分子化合物在溶液中可以离解成离子,称为高分子电解质。其中最重要的就是蛋白质。蛋白质的一个重要特点是,同时具有多个羧基-COOH(在水溶液中可以给出H+离子)和多个氨基-NH2(可接受H+离子),因此蛋白质是一种两性大分子电解质。如果蛋白质在水溶液中分子里的羧基给出的质子被氨基夺去,就形成同时带有正负电荷的两性离子。
[等电状态]蛋白质处于正、负电荷相等时的状态。
[等电点]蛋白质处于等电状态时的水溶液的pH值。一般蛋白质在等电点时pH都不等于7。蛋白质溶液的性质与pH有很大关系,在等电点时蛋白质最不稳定,因为等电状态时蛋白质微粒总电荷为零,水化程度低,容易合并,蛋白质的其它性质如渗透压、导电性、粘度等在等电点也表现得最低或最小。
[表面活性物质]能显著降低水的表面张力的物质称为表面活性物质或表面活性剂。表面活性剂的一般分类是分离子型和非离子型。离子型表面活性剂又有三种:
⑴阴离子表面活性剂,如普通肥皂和洗衣粉(烷基磺酸盐)。
⑵阳离子表面活性剂,如十八胺盐酸盐和十二烷基二甲基苄基氯化铵等。
⑶两性表面活性剂,指同时具有阴离子和阳离子的表面活性剂,如氨基酸、氨基磺酸盐等。
⑷非离子型表面活性剂指在溶液中不电离,不生成离子的表面活性剂,如聚乙二醇型表面活性剂。
[乳化剂]乳浊液(或称乳状液)的稳定剂。乳浊液是热力学不稳定体系,要使它稳定就必需加入乳化剂,其作用是在分散相液滴的表面上形成界面膜或双电层,可以阻止小液滴的相互凝结。很多乳化剂是表面活性物质,如各类肥皂;也有些乳化剂不是表面活性物质,如Al(OH)3、 SiO2、陶土及石墨等固体粉末,由于它们能在相界面形成较坚固的界面膜,也可以形成稳定的乳状液。
[土壤胶体]土壤中呈胶体状态的物体。土壤胶体分有机胶体(如腐植质)、无机胶体(粘土矿物,如铝硅酸盐及铁、铝的含水氧化物等)、有机无机复合胶体三种。土壤胶体具有很大的表面积、在溶液中带有电荷,并有吸收、膨胀、收缩、分散、凝聚、粘结、粘着和可塑等特点。有机胶体吸收性较强,无机胶体则粘结性、可塑性较强,各种胶体在土壤中的存在量,对土壤性状和肥力都有很大影响。
[电解质]溶于水或熔融状态下能导电的化合物。离子化合物和许多极性键化合物都是电解质。如酸、碱、盐等类物质。
[非电解质]一般系指在熔融状态或在水溶液中不能导电的化合物。非电解质都是共价化合物,大多数有机物及许多无机化合物,如非金属卤化物等。(但许多具有共价键的无机化合物溶于水则形成电解质溶液,如卤化氢。)
[电离]通常指电解质在水溶液中或熔融状态下,离解成阴、阳离子的过程。此外,气态分子或原子变为离子的过程也叫电离。
[强电解质]在水溶液中能完全电离的电解质。其水溶液具有较强的导电性,包括典型的离子键化合物及在水分子作用下能完全离子化的极性键化合物。强酸、强碱及大多数盐属于强电解质。
[弱电解质]在水溶液中只能部分电离的电解质称弱电解质。弱电解质在水溶液中,主要以分子状态存在,导电能力较弱,如弱酸、弱碱和几种汞盐及镉盐等。
[强酸]属于强电解质的酸类。如盐酸HCl、硝酸HNO3、硫酸H2SO4、高氯酸HClO4等。
[强碱]属于强电解质的碱类。如氢氧化钠NaOH、氢氧化钾KOH等。
[弱酸]属于弱电解质的酸类。如碳酸H2CO3、氢硫酸H2S、氢氰酸HCN等。
[弱碱]属于弱电解质的碱类。如一水合氨NH3·H2O、氢氧化铁Fe(OH)3等。
[电离平衡]在水溶液中弱电解质分子和离子间的平衡。例如醋酸的电离平衡:
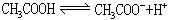
[电离度]电离平衡时,在弱电解溶液中已电离的分子数占电离前分子总数的百分率。它可以表示弱电解质电离的程度,其算式如下:
电离度(a)=已电离的分子数/电离前分子总数×100%
不同的弱电解质,在相同浓度时,它们的电离度不同。电解质愈弱,电离度就愈小,因此,可用电离度表示弱电解质的相对强弱。同一电解质溶液,浓度愈小,电离度愈大。
[电离平衡常数]弱电解质在水溶液中存在电离平衡,跟化学平衡一样也可得到电离平衡常数,用Ki表示(弱酸的电离常数用Ka表示、弱碱的电离常数用Kb表示)。例如醋酸的电离平衡:
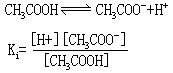
Ki的值与温度有关,而与浓度无关。一定温度下某弱电解质的Ki是定值,Ki越大表示电离程度越大。对同类型的弱酸或弱碱,比较其Ka或Kb值的大小,就可确定它们的相对强弱。如氢氟酸(HF)和氢氰酸(HCN)都是一元酸,前者电离常数Ka=6.5×10-4、后者的电离常数Ka=4.93×10-10,可知氢氟酸比氢氰酸强。
[阿累尼乌斯理论]即阿累尼乌斯电离学说。1887年由瑞典物理化学家阿累尼乌斯(Arrhenius)提出。其要点如下:电解质在水溶液中部分离解为自由移动的离子,即发生电离;溶液愈稀电离度愈大;电离过程是可逆的,分子电离成离子,离子又相互碰撞结合成分子,最后达到电离平衡。电离学说的提出揭示了电离平衡的实质,在此学说的基础上建立了酸碱理论。
[酸碱电离理论]阿累尼乌斯从他的电离学说的观点出发,把在水中能电离出氢离子的化合物称为酸,把在水中能电离出氢氧离子的化合物称为碱。如HCl、H2SO4、CH2COOH等都是酸;NaOH、KOH、Ca(OH)2等都是碱。根据这一理论,酸碱中和反应的实质就是酸电离产生的H+离子和碱电离产生的OH-离子,相互结合生成水的过程。如盐酸和氢氧化钠的中和
HCl+NaOH=NaCl+H2O
其离子方程式为:
H++OH-=H2O
酸碱电离理论对化学科学的发展起了重大作用,但因把酸碱限制在水溶液中而有一定的局限性。以后酸碱理论又有了很大发展。
[稀释定律]对某弱电解质的稀溶液而言,此种弱电解质的电离度和溶液浓度的平方根成反比。其数学表达式为:
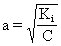
式中a为弱电解质的电离度,Ki为其电离常数,C为溶液的摩尔浓度。确切地说上式表达的只是一种近似关系。
[同离子效应]在弱电解质溶液中,加入与弱电解质具有相同离子的强电解质,而使弱电解质电离度减小的效应。例如浓度为0.1mol/l的醋酸(CH3COOH)溶液的电离度是1.33%,若加入醋酸钠(CH3COONa)晶体使溶液中醋酸钠浓度为0.1mol/l,此时醋酸的电离度降为0.018%。这是由于醋酸钠是强电解质在溶液中电离出大量的CH3COO-离子,使醋酸的电离平衡向形成醋酸分子一方移动的结果。在同离子效应发生的同时,也存在着盐效应(见盐效应),但盐效应对电离度的影响比同离子效应要小得多。
[盐效应]在弱电解质溶液中,加入其它强电解质盐时,使该弱电解质电离度增大的效应。例如0.1mol/l的醋酸溶液,醋酸的电离度为1.33%,若加入氯化钠,使溶液中NaCl的浓度为0.1mol/L,此时醋酸的电离度为1.68%。这是因为加入不含相同离子的强电解质,增大了溶液中离子的总浓度,使溶液中离子之间的相互牵制作用增强,离子结合为分子的机会减少,降低了分子化的速度,电离平衡向电离方向移动的结果。
在难溶电解质的沉淀溶解平衡体系中,加入可溶性强电解质而使沉淀进一步溶解的现象,也称为盐效应。
[水的离子积常数]简称水的离子积。纯水本身能够产生微弱的电离,存在着电离平衡:
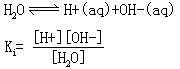
对于纯净的水,其浓度为常数,因此Ki[H2O]也是常数,在298.15K时纯水中[H+]=[OH-]=1.004×10-7摩尔/升所以,在101325Pa,298.15K时,
Kw=Ki[H2O]=[H+][OH-]=(1.004×10-7)2≈10-14
Kw即水的离子积。水的离子积反映了在一定温度下水中H+离子和OH-离子的浓度间的关系。在任何水溶液中,水和它的离子(H+,OH-)之间的平衡是一种非常重要的平衡。在稀溶液中水的浓度可近似地看成为常数(近似等于纯水的浓度55.55摩尔/升),故在任何一种稀溶液中H+离子和OH-离子的浓度关系都服从水的离子积常数公式,[H+][OH-]=10-14。通过Kw,已知H+离子(或OH-离子)的浓度,就可计算出OH-离子(或H+离子)的浓度。
[pH值]水溶液中H+离子浓度的负对数。即
pH=-lg[H+]
pH值在0-14之间。
当[H+]=1摩尔/升时,pH=0;
[H+]=10-14摩尔/升时(此时[OH-]=1摩尔/升),pH=14;
[H+]=[OH-]=10-7摩尔/升时,pH=7。
根据pH值的大小可以判断溶液的酸碱性,pH=7是中性溶液,pH<7是酸性溶液,pH>7是碱性溶液。pH值通常用于表示浓度较小的溶液的酸碱度,当溶液中氢离子的浓度较大时,用pH值表示酸碱度反而不方便。
[缓冲作用]溶液能抵抗外加少量强酸、强碱或稀释的影响,而本身的pH值不发生显著变化的作用。
[缓冲溶液]能抵抗外加少量强酸、强碱或稀释的影响,而本身的pH值不发生显著变化的溶液。缓冲溶液有三种类型:弱酸及其盐(如HAc-NaAc)、弱碱及其盐(NH3·H2O-NH4Cl)、多元弱酸盐及其对应的次级盐(NaH2PO4-Na2HPO4),总之缓冲溶液由弱酸及其共轭碱组成(参见共轭酸碱条)。缓冲溶液具有缓冲能力的原因,以HAc-NaAc混和溶液为例:
HAc H++Ac-
H++Ac-
NaAc=Na++Ac-
当加入少量酸时,体系中Ac-与加入的H+结合成HAc分子,而体系中[H+]无明显增加,Ac-为体系中抗酸部分;当加入少量碱时,体系中的HAc能与之中和,体系中[OH-]亦无明显增加,HAc为体系中的抗碱部份。缓冲溶液的pH值(或pOH值)可按下式计算:
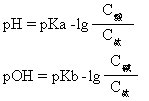
缓冲溶液的缓冲能力是有限的,一般说C酸(或C碱)和C盐的数值较大且浓度相近时能力最大,一般取C酸/C盐或C碱/C盐之值在0.1-10之间。缓冲溶液在工业、农业、医学、化学、生理学方面有重要用途,如土壤、人的血液中都有复杂的缓冲体系,从而保持一定的pH值,使植物正常生长,使人维持正常的生理活动。
[缓冲容量]衡量缓冲溶液缓冲能力的标准。通常指1升缓冲溶液的pH值改变1单位时所需加入强酸或强碱的摩尔数。缓冲容量的大小与缓冲溶液的总浓度及组分比有关,总浓度愈大,且缓冲组分的浓度比愈接近1∶1,缓冲容量愈大。
[盐类的水解]盐的离子与溶液中水电离出的H+离子或OH-离子作用产生弱电解质的反应。由于形成盐的酸和碱的强弱不同,各种盐类水解进行的程度也各有差别。强酸强碱盐不发生水解;弱酸强碱盐水解溶液显碱性;弱碱强酸盐水解溶液显酸性;弱酸弱碱盐水解程度较大,溶液的酸碱性取决于水解产物的相对强弱。
[水解度]在一定温度下,盐溶解在水中,已经水解的盐的浓度与盐的起始浓度的比值,算式如下(水解度用h表示):

水解度的大小,主要决定于盐的本性,同时也受浓度、温度等因素的影响。对于同一种盐的溶液,降低浓度或升高温度可增大水解度。水解平衡也服从化学平衡规律,因此在盐溶液中加入一种与水解产物相同的物质可抑制水解,降低水解度。
[水解常数]水解平衡也是一种化学平衡,其平衡常数即水解常数。例如:
NaAc(强碱弱酸盐)的水解常数:
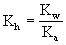
NH4Cl(强酸弱碱盐)的水解常数:
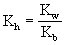
NH4Ac(弱酸弱碱盐)的水解常数:
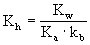
以上各式中Kh为水解常数,Kw为水的离子积,Ka为弱酸电离常数,Kb为弱碱电离常数。
多元弱酸盐的水解,以Na2CO3为例,水解是分步进行的:
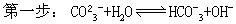
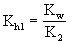
(式中Kh1为第一步水解常数,K2为该酸的二级电离常数)
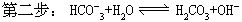
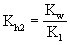
(式中Kh2为第二步水解常数,K1为碳酸的一级电离常数)。因为Kh1》Kh2,所以第一步水解是主要的。
水解常数与水解度有如下关系:
弱酸强碱盐
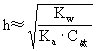
弱碱强酸盐
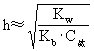
弱酸弱碱盐
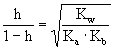
[酸碱质子理论]简称质子理论或质子论。1923年由丹麦的布朗斯特(Br nsted)和美国的劳莱(Lowry)提出。质子理论认为,凡能给出质子(H+)的物质都是酸,凡能接受质子的物质都是碱。如HCl、NH4+、HSO4-等能给出质子,都是酸;Cl-、NH3、SO42-等能接受质子,都是碱。根据质子理论,酸和碱不是孤立的,它们存在着如下的关系:
nsted)和美国的劳莱(Lowry)提出。质子理论认为,凡能给出质子(H+)的物质都是酸,凡能接受质子的物质都是碱。如HCl、NH4+、HSO4-等能给出质子,都是酸;Cl-、NH3、SO42-等能接受质子,都是碱。根据质子理论,酸和碱不是孤立的,它们存在着如下的关系:
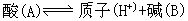
这种对应关系叫共轭关系,称共轭酸碱对,右边的碱(B)是左边的酸(A)的共轭碱;而邹边A是B的共轭酸。例如在NH4+ NH3+H+中,NH4+和NH3就是一对共轭酸碱。由此可见,质子论不是从组成上区分酸碱,而是看这种物质(分子或离子)在反应中对H+的授受关系。如HCO3-在HCO3--CO32-的共轭关系中是酸,而在H2CO3-HCO3-共轭关系中是碱:
NH3+H+中,NH4+和NH3就是一对共轭酸碱。由此可见,质子论不是从组成上区分酸碱,而是看这种物质(分子或离子)在反应中对H+的授受关系。如HCO3-在HCO3--CO32-的共轭关系中是酸,而在H2CO3-HCO3-共轭关系中是碱:
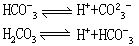
据质子理论,酸碱反应的实质就是两个共轭酸碱之间质子传递的反应,如HCl和NH3的反应:
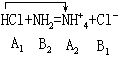
酸碱质子理论,扩大了酸碱的范围,是酸碱理论的发展,可以把电离理论中的电离、中和、水解、同离子效应等都包括在酸碱反应的范围内,看作是质子传递的酸碱反应。
电离,酸碱在水溶液中的电离,可看成它们与水分子间的质子传递,例如:
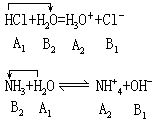
在酸碱质子反应中,水既可以接受质子是碱,又可以给出质子是酸。
中和,电离理论中的酸碱中和也是质子传递作用:
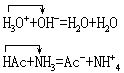
水解,质子理论中没有盐的概念,水解反应相当于质子理论中离子酸(或离子碱)与水之间的质子传递反应,例如:
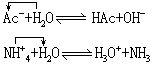
[离子酸]酸碱质子理论中所说的酸(见酸碱质子理论),若是离子则称离子酸,若是分子则称分子酸。离子酸又分阳离子酸,如NH4+(NH4+=H++NH3);以及阴离子酸,如HCO3-(HCO3-=H++CO32-)。
[离子碱]酸碱质子理论中所说的碱(见酸碱质子理论),若是离子则称离子碱,若是分子则称分子碱。离子碱又分阳离子碱,如Fe(OH)2+;以及阴离子碱,如CO32-。
[共轭酸碱]见酸碱质子理论条。
[路易斯酸碱理论]又称酸碱电子理论。1923年由美国化学家路易斯(G.N.Lewis)提出。其要点是:凡是可以接受电子对的物质称为酸,凡是可以给出电子对的物质称为碱。故酸又称为电子对接受体,碱又称电子对给予体。按照路易斯的观点,酸碱反应的实质是配位键的形成并生成酸碱络合物。例如:

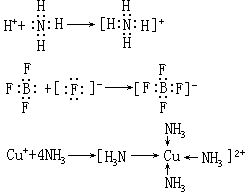
路易斯酸、碱的范围非常广泛,酸碱络合物包含很多类物质。凡是金属离子及氢离子都是酸,与路易斯酸结合的物质不管是阴离子或中性分子都是碱。所以大多数无机化合物都可看成是酸碱络合物,许多有机化合物也是如此。酸碱电子理论以电子对的授受来说明酸碱反应,更能体现物质的本质属性。但由于对酸碱的认识过于笼统,没有统一的酸碱强度的标准,因而有一定的局限性。
[电子对接受体]见路易斯酸碱理论条。
[电子对给予体]见路易斯酸碱理论条。
[路易斯酸]见路易斯酸碱理论条。
[路易斯碱]见路易斯酸碱理论条。
[软酸]路易斯酸中具有体积大、正电荷少、易极化、易失去电子、易形成共价性较强的键的物质。如Cu+、Ag+、Hg22-、Cd2+等属于软酸。软酸易与软碱生成稳定的络合物,软酸不易与硬碱生成络合物,或生成的络合物不稳定。
[软碱]路易斯碱中,具有电负性小、易失去电子、其低能轨道不饱和、易形成共价性较强的键的物质,如I-、CN-、S2O32-等属于软碱。软碱易与软酸生成稳定的络合物,软碱与硬酸不易生成络合物,或生成的络合物不稳定。
[硬酸]路易斯酸中具有体积小、正电荷多、不易失去电子、不易极化、易形成离子性较强的键的物质,如Li+、Na+、Ca2+、Mg2+、Fe3+等属于硬酸。硬酸易与硬碱生成稳定络合物,硬酸与软碱不易生成络合物,或生成的络合物不稳定。
[硬碱]路易斯碱中具有电负性大、不易极化、不易失去电子,而且低能轨道已饱和、易形成离子性较强的键的物质,如H2O、Cl-、OH-等属于硬碱。硬碱易与硬酸生成稳定的络合物,硬碱与软酸不易生成络合物或生成的络合物不稳定。
[沉淀溶解平衡]在溶液中,难溶电解质的溶解过程与其相应的离子结合为沉淀的过程之间的动态平衡。此时溶液已达到饱和,这是难溶电解质的饱和溶液中离子与未溶固体物质间的多相平衡。如难溶电解质AgCl的沉淀溶解平衡:
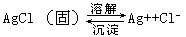
[溶度积常数]即难溶电解质在水溶液中达到沉淀溶解平衡时的平衡常数。例如:
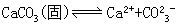
因CaCO3是固体,其浓度是一定值,平衡常数表达式为:
Ksp=[Ca2+][CO32-]
Ksp就是溶度积常数,简称溶度积,它表示在难溶电解质的饱和溶液中,有关离子浓度的乘积在一定温度下是个常数。根据溶度积常数值可比较同类型的难溶电解质溶解度的大小,溶度积越小溶解度也越小。
[溶度积规则]用溶度积概念判断在一定条件下,某难溶电解质在溶液中沉淀能否生成或溶解的规则。在某难溶电解质溶液中,其离子浓度的乘积称为离子积,用Qi表示。在给定的溶液中Qi可能有三种情况:
⑴Qi=Ksp是饱和溶液
⑵Qi<Ksp是不饱和溶液,无沉淀析出,若体系中已有固体存在,沉淀会溶解,直至饱和(Qi=Ksp)为止。
⑶Qi>Ksp是过饱和溶液,有沉淀析出,直至饱和。
以上就是溶度积规则。溶度积规则有一定的局限性,如形成的难溶固体很少(少于每毫升10-5克),或形成过饱和溶液以及产生副反应时,虽然Qi已大于Ksp,也观察不到沉淀的生成。
[分步沉淀]在含有几种离子的溶液中,加入一种沉淀剂,根据溶度积规则,对同类型的难溶化合物,Ksp小的先沉淀,Ksp大的后沉淀,这种先后沉淀作用叫分步沉淀。例如在0.1mol/L的Cl-和I-溶液中逐滴加入AgNO3溶液,先生成AgI沉淀,后生成AgCl沉淀。利用分步沉淀的原理可将两种离子分离。沉淀的先后顺序除跟溶度积有关外,还与被沉淀离子的起始浓度有关。
[强电解质理论]又称德拜-休格尔理论。是1923年由物理化学家德拜(Debye)和休格尔(Hückel)提出。其要点是:强电解质在溶液中是完全电离的,但溶液中离子间又存在着一定的静电相互作用,溶液中的离子都被具有相反电荷的离子所包围,形成“离子氛”,因而影响到离子的运动速度和溶液的性质。这一理论解释了强电解质完全电离而其表观电离度又不是100%的矛盾。
[德拜-休格尔理论]见强电解质理论条。
[离子互吸理论]见强电解质理论条。
[表观电离度]系指通过实验测得的强电解质的电离度。强电解质的电离度和弱电解质的电离度的意义不同,强电解质的电离度反映了溶液中离子间相互牵制作用的强弱程度。
[活度]指电解质溶液中表观上某离子的浓度,又称有效浓度或校正浓度。它与实际浓度的关系是:
a=r·c
式中a为某离子的活度,c为该离子的实际浓度,r为活度系数(也有用f表示的)。活度表明体系与理想状态的偏差。
[活度系数]溶液的实际浓度除活度所得之商。对于理想溶液,r=1,此时a=c,活度等于浓度。在一般情况下r<1,且随浓度而变化,溶液浓度愈大,离子之间的牵制作用愈强,活度与浓度之间的差距愈显著;溶液浓度愈小,离子之间的牵制作用愈弱,活度与浓度之间的差距愈不显著,当溶液极稀时,由于离子间的相互作用变得十分微弱,而r趋近于1,活度与浓度趋于相等。
[有效浓度]见活度条。
[无限稀释]有两种含义:
⑴指溶液的浓度趋近于零的状态,在无限稀释时,所有的电解质全部电离,且离子间一切相互作用均可忽略。
⑵指溶液稀释到再加溶剂时无热效应的状态。
[电导]描述物体导电性能的物理量,其值是电阻的倒数,单位是“西门子”,用S表示。某物体的电阻愈小它的电导就愈大。
[电导率]又称比电导。可用来衡量和比较物体的导电性能。当物体的长度为1厘米,截面积为1平方厘米时的电导,为该物体的电导率。对电解质溶液来说,当两电极面积各为1平方厘米,电极相距1厘米时为溶液的电导。电解质溶液的电导率与电解质的种类、溶液的浓度及温度等有关。
[离子淌度]又称离子迁移率。某种离子在一定的溶剂中,当电位梯度为每米1伏特时的迁移速率称为此种离子的淌度,单位是米2·秒-1·伏特-1。离子淌度是代表离子迁移速率特征的物理量。
[离子迁移率]见离子淌度条。
[离子强度]在电解质溶液中离子之间平均静电相互作用的一种量度,常用I表示,其定义式如下:
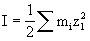
上式的含义是:某电解质溶液的离子强度等于每种离子的质量摩尔浓度mi与其价数Zi平方的乘积之和的一半。
[双电层]在两种不同物体的接触界面上,正负电荷分别排列成的一种面层,例如在金属和电解质溶液相接触的界面上,金属表面电荷层与液溶中的相反电荷的离子层就构成双电层,由于溶液中的离子的热运动,溶液中的一层又分为紧密层和扩散层。
[可逆电池]热力学意义上的可逆电池必须具备两个条件,即可逆电池在充电和放电时不仅物质的转变是可逆的,而且能量的转变也是可逆的。如果把放电时放出的电能全部储存起来并用它对电池充电,则能恰好使电池内化学反应体系及外界环境全部恢复原状。
一般意义的可逆电池就是指由于放电而被消耗的物质,可以通过充电使之再生的电池,此电池的充电反应恰好是放电反应的逆过程,如常用的铅蓄电池。
[可逆电极]构成可逆电池的电极。主要有以下三种类型:
⑴金属电极、氢电极、氧电极、卤素电极。金属电极是将金属浸在含有该种金属离子的溶液中构成,以符号M|M2+表示;氢电极、氧电极、氯电极,分别是将被H2、O2、Cl2的气流冲击着的铂片浸入含有H+、OH-、Cl-的溶液中构成,分别用符号(Pt)H2|H+或(Pt)H2|OH-、(Pt)O2|OH-或(Pt)O2|H2O,H+以及(Pt)Cl2|Cl-表示。
⑵微溶盐电极和微溶氧化物电极。微溶盐电极是将金属覆盖一薄层该金属的一种微溶盐,然后浸入含有该微溶盐负离子的溶液中构成,如银-氯化银电极,Ag-AgCl|Cl-;微溶氧化物电极是将金属覆盖一薄层该金属的氧化物,然后浸在含有H+或OH-的溶液中构成,如汞-氧化汞电极,Hg-HgO|OH-。
⑶氧化还原电极,由惰性金属如铂片插入含有某种离子的两种不同氧化态的溶液中构成,如Fe2+-Fe3+电极,(Pt)|Fe2+,Fe3+。
[半电池]每个可逆电极都可看成是一个半电池。
[盐桥]如果在一个电池中存在着两个电解质溶液的接界面,则在界面处也产生一个电位差,这叫液体接界电位。盐桥是消除液体接界电位的一种装置。两个半电池在构成电池时,电解质溶液不直接接触,而用盐桥沟通,盐桥是用浓KCl溶液和3%琼脂凝聚而成,由于K+和Cl-的迁移数很相近,在盐桥两侧形成两个数值小又几乎相等且符号相反的液体接界电位,使净接界电位降低到只有1-2毫伏,可以忽略不计。如果电池中电解质溶液能与KCl反应(如含Ag+),可采用KNO3或NH4NO3代替KCl做盐桥。
[原电池]通过氧化还原反应而产生电流的装置,也可以说是把化学能转变成电能的装置。有的原电池可以构成可逆电池,有的原电池则不属于可逆电池。
[标准电池]是一种电动势比较稳定,能保持长期不变的可逆电池。标准电池用来测定或校正电池的电动势。常用的是韦斯登(Westone)标准电池,它的正极是汞(Hg)和硫酸亚汞(Hg2SO4)的糊状物,下方放少量汞为与引出的导线保持良好的接触;负极是镉汞齐(Cd-Hg),在糊状物和汞齐上方分别放有CdSO4·83H2O晶体和它的饱和溶液。电极反应和电池反应分别为
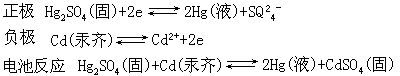
这种电池的电动势很稳定,20℃时Es=1.01865伏,温度对这种电池的电动势的影响很小。
[标准氢电极]把镀有铂黑的铂片放到氢离子浓度为1摩尔/升(严格地说应是活度aH+=1)的酸溶液中,在25℃(298.15K)时以pH2=101325帕(1大气压)的干燥氢气不断冲击到铂电极上,使铂黑上吸附的氢气达到饱和,这就构成了标准氢电极。规定在任何温度下标准氢电极的电极电位为零。标准氢电极是常用的参比电极,其它电极的电极电位是和标准氢电极相比较而得到的数值。
[电极电位]将标准氢电极作为发生氧化作用的负极,而将待定某电极作为发生还原作用的正极,组成一个电池。(Pt)H2(pH2=101325Pa)|H+(aH+=1)||待定电极这个电池电动势的数值和符号就是待定电极电位的数值和符号,可以看出,电极电位是一相对值。通常把电极-溶液界面的电位差称为绝对电极电位。
[标准电极电位]又称标准电极电势,通常用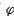 表示。以标准氢电极作为负极,某给定电极为正极组成电池,若此给定电极处于标准状态,即组成电极的离子浓度为1摩尔/升(严格地说,应是活度为1),气体的分压为101325帕(1大气压),液体和固体为纯态,此时电池的电动势就是给定电极的标准电极电位。标准电极电位的数值越大(正值越大),说明该电对中的氧化态的氧化性越强,标准电极电位的数值越小(负数的绝对值越大),说明该电对中的还原态还原性越强。标准电极电位有广泛的应用,例如可以判断氧化-还原反应的方向和程度,可以求算水溶液中发生的氧化还原反应的化学平衡常数,可以求算难溶盐的溶度积等。
表示。以标准氢电极作为负极,某给定电极为正极组成电池,若此给定电极处于标准状态,即组成电极的离子浓度为1摩尔/升(严格地说,应是活度为1),气体的分压为101325帕(1大气压),液体和固体为纯态,此时电池的电动势就是给定电极的标准电极电位。标准电极电位的数值越大(正值越大),说明该电对中的氧化态的氧化性越强,标准电极电位的数值越小(负数的绝对值越大),说明该电对中的还原态还原性越强。标准电极电位有广泛的应用,例如可以判断氧化-还原反应的方向和程度,可以求算水溶液中发生的氧化还原反应的化学平衡常数,可以求算难溶盐的溶度积等。
[氧化-还原电对]氧化-还原反应可看成是两个半反应的总和,在每个半反应中氧化型物质和还原型物质组成一个氧化-还原电对。例如
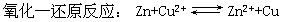
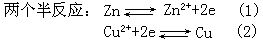
在半反应⑴中,氧化型物质Zn2+和还原型物质Zn组成一个氧化-还原电对Zn2+/Zn;在半反应⑵中,氧化型物质Cu2+和还原型物质Cu组成一个氧化-还原电对Cu2+/Cu。实际上组成可逆电极的物质都是一个氧化-还原电对(见可逆电极)。
[参比电极]在测定某电极的电极电位时,必须与一个已知电极电位的电极构成一个电池,测定出此电池的电动势后,就可以计算出被测电极的电极电位。这种已知电极电位的电极,称为参比电极。如氢电极、甘汞电极、银-氯化银电极等。
[甘汞电极]是常用的一种参比电极。由汞和氯化亚汞(Hg2Cl2)在氯化钾水溶液中的饱和溶液相接触而成。常用的甘汞电极有三种:氯化钾溶液为饱和溶液的是饱和甘汞电极、氯化钾溶液浓度为1摩尔/升的是当量甘汞电极、氯化钾溶液浓度为0.1摩尔/升的是0.1摩尔/升甘汞电极。在298.15K时,当量甘汞电极的电极电位是0.2801伏。甘汞电极的制备和保存都很方便,电极电位很稳定,所以用途很广。
[氢醌电极]由铂(或金)电极和醌氢醌的水溶液构成。醌氢醌是醌和氢醌以等摩尔数相结合的物质,是墨绿色晶体,分子式为C6H4O2·C6H4(OH)2。这种电极用于测定溶液的pH值,其优点是操作简单,只需将少量醌氢醌加入pH待测液中(因醌氢醌溶解度很小),再插入一支光亮的铂电极,就构成了一个醌氢醌电极。这种电极在碱性溶液和浓盐溶液中不宜使用。
[玻璃电极]用于测定溶液pH值的电极。将一种特制的玻璃制成小球,球内盛pH值为定值的缓冲溶液,用铂丝做导线就构成玻璃电极。将玻璃电极浸入待测液中和一参比电极(通常用甘汞电极)组成电池,通过测定电池的电动势就可以算出待测液的pH值。用玻璃电极测定溶液pH值的装置叫pH计。玻璃电极具有不受溶液中氧化性物质及各种杂质影响的特点,所需待测液的数量也很少,操作简便,所以得到了广泛的应用。
[金属活动性顺序]目前中学化学教科书中按金属在水溶液中形成低价离子(指在水溶液中能比较稳定存在的简单离子)的标准电极电位顺序排列的。现行教材的金属活动性顺序是:

金属活动性顺序只是从热力学的角度指出了发生氧化-还原反应的可能性、趋势大小,而不能说明其反应的快慢。金属活动顺序是金属在标准状态下(金属为纯净的,金属离子在溶液中活度为1)的活动性顺序,是金属在水溶液中形成简单离子的活动性顺序,若介质改变活动性会有变化。在运用金属活动性顺序时,必须根据具体条件进行分析,才能得出正确的结论。
[能斯特方程]由德国物理化学家能斯特(Nernst)提出的对任何一种电池反应的电池电动势与电池本性和电解质浓度间的定量关系式,如:
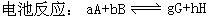
电池电动势:
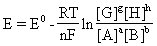
式中E为一定状态下电池的电动势,E0为该电池在标准状态下的电动势,即标准电动势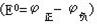 ,R为气体常数,T为绝对温度,n为电子转移的摩尔数,F是法拉第常数96.487千焦/伏·摩尔。
,R为气体常数,T为绝对温度,n为电子转移的摩尔数,F是法拉第常数96.487千焦/伏·摩尔。
当温度为298.15K时,能斯特方程为:
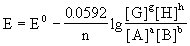
对某电极的电极电位的能斯特方程为:
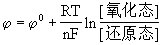
当温度为298.15K时为:
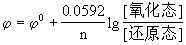
[电势-pH图]在许多电极反应(即氧化-还原反应)中,H+或OH-的氧化数虽然没有变化,却参与了电极反应,它们的浓度对电极电位也有影响。以pH值为横座标,以电极电位为纵坐标,绘出电极电位随pH值变化的关系图,称为电势-pH图。例如水的电势-pH图,有两条线,位于下方的一条线称为氢线,表示水被还原放出氢气时,电极电位随pH值的变化;与氢线平行位于上方的一条线称为氧线,表示水被氧化放出氧气时,电极电位随pH值的变化。
[元素标准电势图]表明某一元素的各种氧化态之间标准电极电势变化关系的图。不同的氧化态按由高向低顺序排列,线上的数字是电对的标准电极电势,酸性介质中的电极电势用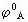 表示,碱性介质中的电极电势用
表示,碱性介质中的电极电势用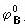 表示。例如铁元素在酸性介质中的电极电势图:
表示。例如铁元素在酸性介质中的电极电势图:
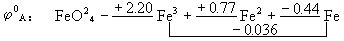
元素标准电势图有广泛的应用,主要有:
⑴从已知电对求未知电对的标准电极电势,计算公式为
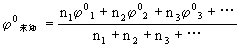
例如已知酸性介质中氯元素的标准电势图:
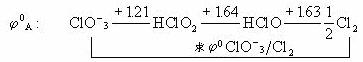
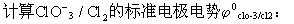
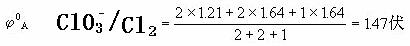
⑵判断某元素的中间价态能否发生歧化反应,如下列元素电势图中A为高价态,B为中间价态,C为低价态:
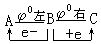
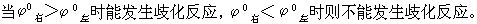
[电极的极化]在电流通过电极时,电极电位偏离平衡值的现象,称为电极的极化。电极极化在现实的电化学过程中是普遍存在的,主要有浓差极化和活化极化。
[过电位]为了表示出电极极化的状况,把在某一电流密度下极化电极电位与该电极的平衡电极电位之间的差值称为过电位(以η表示),η都取正值,则
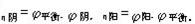
[分解电压]电解时能使电流继续稳定地通过电解质,并使之开始电解的最低电压。分解电压的大小和电极性质,电解液组成、温度、电流密度等因素有关。
[超电压]电解时,实际所需的分解电压超过理论值(指两电极的可逆电动势)的那部分电压称为超电压。在一般情况下,气体的超电压都比较大,金属的超电压(除Fe、Co、Ni外)都比较小。对同一析出物来说,超电压也不恒定,如氢气在铁电极上析出时超电压较小,而在镍电极上析出时超电压就较大。在电解中超电压的存在造成多耗电能,是不利因素,但有时也能加以利用,如在微酸性的锌盐溶液中镀锌时,由于氢气的超电压较大,实际上在镀件上析出的是锌而不是氢气。
[法拉第电解定律]1833年英国物理学家法拉第(Fara day)发现;其要点是:
⑴电解时在电极上电解产物的量与溶液通过的电量成正比;
⑵当以相同的电量分别通过不同的电解质溶液时,在各电极上析出或溶去的物质具有相同的当量值。
现代关于电解定律的叙述是:1法拉第的电量(1摩尔电子的总电量是96487库仑,称为1法拉第,用F表示),可以析出或溶解相当于接受或给出1摩尔电子的物质。
[电解]即在直流电的作用下,电解质所发生的氧化-还原反应。电解池的阳极与电源的正极相连,电解池的阴极与电源的负极相连,通电时阳极发生氧化反应,阴极发生还原反应。电解有着重要的应用,许多基本化工产品的生产、稀有金属的冶炼、金属的精制、电镀等都要利用电解的方法。
[电镀]应用电解原理在金属制品或塑料制品表面镀上一层其它金属或合金的方法。电镀的目的主要是使金属增强抗腐蚀性、增加美观和制品表面硬度。镀层的金属或合金通常有:铬、锌、镍、银、铜锌合金等。电镀时,镀件为阴极,所镀的金属或合金为阳极,含有镀层金属离子的溶液为电解液。
[阳极氧化]利用电解方法使金属制品表面形成附着力很强的氧化物薄膜的过程。这层薄膜可防止金属的腐蚀,并可做金属着色的基底。此外,在电化学反应中,在阳极上发生的氧化反应有时也叫阳极氧化。
[化学电源]把化学能转变为电能并可做为电能来源的装置叫化学电源。日常使用的干电池、蓄电池等都是化学电源。
[干电池]指锌锰干电池,是常用的化学电源之一,是一次电池。负极是锌,正极是被二氧化锰包围着的石墨电极,电解质是氯化锌及氯化铵的糊状物。这种电池应用相当广泛,已有一百多年的历史,但对它的电极反应仍未彻底弄清楚,一般认为电极反应是:
负极 Zn+2NH4Cl-2e→Zn(NH3)2Cl2+2H+
正极 2MnO2+2H++2e→2MnOOH
总反应 Zn+2NH4Cl+2MnO2→Zn(NH3)2Cl2+2MnOOH
这种电池在开路时电压为1.5伏。
[蓄电池]指铅蓄电池,也是常用的化学电源之一,是二次电池。负极是海棉状铅;正极是涂有二氧化铅的铅板,电解质是比重约为1.28的硫酸水溶液。电池表示式为Pb-PbSO4|H2SO4|PbSO4-PbO2-Pb
电极反应和电池反应是
负极 Pb+SO42--2e→PbSO4
正极 PbO2+H2SO4+2H++2e→PbSO4+2H2O
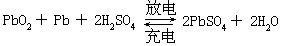
这种电池的电动势为2伏,电池内硫酸的浓度随着放电的进行而降低,当硫酸的相对密度降至约1.05时,电池电动势下降到约1.8伏,应暂停使用,以外加直流电源充电直至硫酸的相对密度恢复到约1.28时为止。蓄电池还有其它类型,如镍铁蓄电池,镍镉蓄电池等。
[银锌电池]一种高能化学电源,可做成蓄电池。负极是锌,正极是氧化银,电解质是40%的KOH溶液和K2ZnO2饱和溶液。电极反应和电池反应是:
负极 Zn+2OH--2e→Zn(OH)2
正极 Ag2O+H2O+2e→2Ag+2OH-

银锌电池具有高的比能量,能大电流放电,因此适用于火箭、导弹和人造卫星、宇航等方面。
[燃料电池]一种高能化学电源,是将燃料和氧气反应时的化学能直接转变成电能的装置。电池的正极和负极都是用微孔导电材料制成(镍、铁、氧化铁等)的。以氢一氧燃料电池为例说明,电池表示式可写成:
(Ni)H2(气)|KOH(20-40%)|O2(气)(Ni)电极反应和电池反应为:
负极 H2+2OH--2e→2H2O
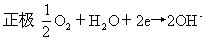
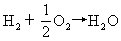
该电池的电动势与氢气和氧气的分压有关。燃料电池的能量利用率很高,氢氧燃料电池已实际应用在宇宙航行和潜艇中,它不仅能大功率供电,而且还具有可靠性高,无噪声以及能供给饮水等优点。
[电化学腐蚀]金属腐蚀的一种普遍形式。由金属接触到电解质溶液构成微电池而发生的金属腐蚀过程叫电化学腐蚀。电化学腐蚀中的微电池,其负极一般都是金属被氧化形成离子进入溶液。
如钢铁在潮湿空气中的腐蚀,Fe-2e=Fe2+。
[析氢腐蚀]电化学腐蚀的一种形式。通常在酸性溶液中、在氢气过电位较小的材料上容易发生。此时在微电池的正极发生的是析出氢气的反应:2H++2e→H2↑。
[吸氧腐蚀]电化学腐蚀的一种形式。通常在中性或碱性溶液中,以及溶液里溶有足够的氧气时发生。此时微电池的正极发生溶解的氧气被还原的反应:O2+2H2O+4e→4OH-。
[阳极保护]为防止金属腐蚀,将被保护的金属作为阳极,在一定条件下进行阳极氧化,使金属钝化(在金属表面形成金属氧化物组成的钝化膜),这种方法叫阳极保护。
[阴极保护]将被保护的金属变为阴极,以防止金属腐蚀的方法。阴极保护法有两种:
⑴外加电流的阴极保护法。把要保护的金属设备作为阴极与外电源的负极相连,另外用不溶性电极作为辅助阳极,与外电源的正极相连,两电极都与电解质溶液接触。通电后,大量电子被强制流向阴极,使金属制品表面产负电荷(电子)积累,只要维持足够的外加电压,由金属腐蚀而产生的原电池的电流就不能被输送,因而金属就可不被腐蚀。这种方法是经济、有效的防腐方法之一,目前在保护闸门、地下贮槽、输油管、电缆及受海水或淡水腐蚀的设备、结晶槽、蒸发罐等多采用这种方法防腐。
⑵牺牲阳极保护法。在要保护的金属设备上联结一种负电位更低的金属,作为更有效的阳极。这样,在发生电化学腐蚀时,被腐蚀的是作为阳极的金属,因此叫牺牲阳极保护法。这种方法可用来防止轮船船壳的腐蚀,通常在轮船的尾部和船壳水线以下部分装上一定数量的锌块作被牺牲的阳极。
[缓蚀剂保护法]是防止金属腐蚀应用得最广泛的方法之一,把少量的缓蚀剂加到腐蚀性介质中,就可使金属腐蚀的速度显著减慢,这种防腐方法叫缓蚀剂保护法。加入缓蚀剂可使微电池的阳极或者阴极(或二者)的极化增加,从而使腐蚀的速度减慢。缓蚀剂的种类很多,无机物有亚硝酸盐、铬酸盐、重铬酸盐、磷酸盐等,有机物有胺类、醛类、杂环化合物、咪唑啉类等。使用缓蚀剂时,要根据被保护的金属种类、腐蚀介质等条件来确定。
[反应速度]化学反应进行的快慢程度用反应速度表示。对于在恒容条件下进行的反应,通常用一种反应物或生成物的浓度在单位时间内的改变来表示反应速度。化学反应的速度是随时间而变化的,因此常用微商的形式表示反应速度。如下列化学反应:
aA+bB→gG+hH
其速度可以分别表示为-dCA/dt、-dCB/dt、-dCG/dt、dCH/dt,因习惯上取反应的速度的数值为正值,而反应物A、B的浓度CA、CB随时间的变化率dCA/dt和dCB/dt本身是负值,所以在表示反应速度时前面应加“-”号。由于化学反应方程式中各化学式的系数之间有一定的关系,所以对同一个反应用不同物质所表示的反应速度应有下列关系:
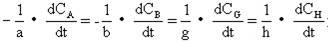
影响反应速度的因素除了反应物的性质外,其它条件,如温度、浓度、压力、催化剂、反应物颗粒的大小等都对反应速度有重要影响。
[反应速度方程式]表示反应速度与浓度间的函数关系式叫做反应速度方程式,又称反应速度公式。某一反应的反应速度方程式只能通过实验找出其具体表达形式。
[基元反应]能代表反应机理的,由反应物微粒(分子、原子、离子、自由基等)一步直接实现的变化,称为基元反应或基元步骤。例如
NO2+CO→NO+CO2
大多数化学反应不是基元反应,有不少是由多个基元反应组成的,例如H2(气)与I2(气)化合的反应是由两个基元步骤完成的:
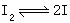
H2+2I→2HI
[简单反应]仅由一个基元步骤完成的反应叫简单反应。
[复杂反应]由多个基元步骤完成的反应叫复杂反应。
[质量作用定律]由挪威化学家葛尔德保(Guldberg)和威吉(Waage)首先提出。原意是:当温度恒定时,在均相系中,化学反应的速度和反应物的有效质量成正比(有效质量应理解为浓度)。现代对质量作用定律的认识是:在恒温下,对简单反应(或复杂反应中的任一基元反应)来说,化学反应的速度和反应物浓度的方次的乘积成正比(反应物浓度方次的数值等于反应式中各分子式前边的系数)。例如有一简单反应的反应式为aA+bB=C,该反应的反应速度方程式为:
v=k[A]a[B]b
式中k为比例常数,又称反应速度常数,在一定的条件下,当所有反应物的浓度都是1mol·1-1时,v=k。
[反应速度常数]反应速度方程式中的比例常数k叫做反应速度常数。例如有以下简单反应:
aA+bB→gG+hH
用A物质浓度变化率表示反应速度时:
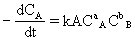
用B物质浓度变化率表示反应速度时:
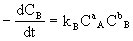
kA与kB有如下关系:kA/kB=a/b。
如果上述反应是气相反应,则反应速度方程式中各物质的浓度可以换成分压,那末式中的k值也要相应地改变。
对不同的反应,k值是不同的。对某指定的反应,k值与反应物的浓度无关而与反应温度及使用的催化剂有关。
[反应级数]反应速度方程式中浓度项的方次之和称为此反应的反应级数。例如对以下反应:
aA+bB→gG+hH
根据实验确定其反应速度方程式为:v=kCaACbB,此反应的反应级数为(a+b),也称该反应为(a+b)级反应。反应级数除有整数级数,如零级、一级、二级、三级等,还可以有分数甚至是负数。反应级数都是由实验测定的。
[反应分子数]指在基元反应过程中参加反应的微粒(分子、原子、离子、自由基等)数目。根据反应分子数可将化学反应区分为单分子反应、双分子反应和三分子反应。例如:
CH3COCH3→C2H4+CO+H2
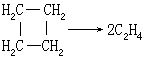
是单分子反应。
CH3COOH+C2H5OH→CH3COOC2H5+H2O
是双分子反应。
H2+2I→2HI
是三分子反应。
大多数反应是单分子反应或双分子反应,三分子反应很少见,至于四分子及四分子以上的反应,从理论上分析几乎是不可能存在的。应该注意,反应分子数和反应级数是两个不同的概念,反应分子数是为了说明反应机理而引出的概念,反应级数是根据实验得出的数值。一般说来,对同一个反应它们的数值是不同的,例如,有零级反应,但没有零分子反应;反应级数可以是分数,反应分子数却不可能是分数。
[可逆反应]又称对峙反应,指在同一条件下,既能向正反应方向进行,同时又能向逆反应方向进行的反应。严格地说,任何反应都是可逆反应。假若逆反应的速度远远小于正反应的速度,平衡点远远偏向产物一边,这种反应可认为是能进行到底的单向反应;假若逆反应的速度相当大,则不能忽略,这类反应就是对峙反应。
[平行反应]当反应物同时进行着两个或两个以上不同的反应时,称为平行反应。这在有机反应中比较普遍。例如甲苯的硝化反应,同时进行着产生邻位、对位、间位硝基甲苯的三个反应。
[连串反应]如果一个复杂反应要经过几个基元反应才能达到最终产物,其中前一个基元反应的产物为后一个基元反应的反应物,如此连续进行,这样的反应,称为连串反应。例如苯的液相氯化:
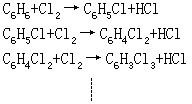
[链反应]这是一类比较特殊的反应。这类反应的特点是:反应一旦开始,如果不加控制,就可发生一系列的连串反应,使反应自动进行下去,故称为链反应。链反应一般分为三个阶段:链的引发、链的传递和链的终止。链的引发需要吸收能量,对反应条件也很敏感,反应一旦引发,通常都进行得非常迅速。例如氢气和氯气化合的反应就是链反应,其反应过程如下:
链的引发:在光照条件下产生自由基(或称活性质点),可表示为
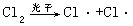
链的传递:自由基参加反应,同时又产生新的自由基,新的自由基又参加反应,再产生新的自由基……不断往下传递,其反应过程是:
Cl·+H2→HCl+H·
H·+Cl2→HCl+Cl·
………………
链的终止:是自由基本身结合为正常分子的步骤。若自由基与器壁或其它分子碰撞耗散了能量而使链终止。
Cl·+Cl·+M(器壁或其它分子)→Cl2+M
根据链反应中链传递的方式不同,又可分为直链反应和支链反应。
[有效碰撞理论]有关反应速度的理论,是在气体分子运动论的基础上提出的。此理论的要点是:在气体反应中,化学反应发生的先决条件是反应物分子必须发生碰撞;但不是任何两种反应物分子之间的碰撞都能发生反应,只有少数分子的碰撞能发生反应,这种能够发生反应的碰撞称为有效碰撞。能发生有效碰撞的分子所具有的能量比整个体系内分子的平均能量要高出很多,才能发生有效碰撞。因此化学反应的速度主要取决于单位时间内有效碰撞的次数。
[有效碰撞]见有效碰撞理论条。
[过渡状态理论]也是一种有关反应速度的理论,是在统计力学和量子力学的发展中建立起来的。该理论的大意是:由反应物到产物必须经过一种过渡状态,即具有足够能量的反应物分子形成活化络合物的中间状态,反应物和活化络合物之间很快达到平衡,化学反应的速度由活化络合物的分解速度来决定。例如A原子和BC分子间的简单反应:
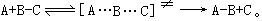
当A原子沿B-C轴线逐渐接近BC分子时,B-C间的化学键逐渐松弛,同时开始逐渐形成新的A-B键,在此过程中体系的位能增加,当形成过渡状态的活化络合物[A…B…C]≠时,体系的位能最高,活化络合物很不稳定,它可能分解变为产物,也可能重新变回反应物。由反应物变成活化络合物及由活化络合物变回反应物的速度都很快,在瞬间就能达到平衡,而由活化络合物分解变成产物的速度却是比较慢的,因此化学反应的速度取决于活化络合物分解的速度。
[活化分子]见有效碰撞理论条。
[活化能]活化能是化学动力学中的一种重要概念,1889年由瑞典物理化学家阿累尼乌斯(Arrhenius)首先提出。阿累尼乌斯总结了大量的实验数据,提出了经验公式
K=Ae-Ea'RT
式中K为速度常数,A为一常数通常称为“指前因子”或“频率因子”,R为气体常数,T为热力学温度,而Ea即为活化能。活化能的意义是:活化分子的平均能量与反应物分子平均能量之差。
在过渡状态理论中,活化能(能垒)是指反应物分子和活化络合物分子处于基态时的位能差,它不等于Ea。
[活化络合物]见过渡状态理论条。
[活化过程]在过渡状态理论中,由稳定的反应物分子过渡到活化络合物的过程叫活化过程。
[阿仑尼乌斯方程式]见活化能条。
[自由基]是一种具有未成对电子的原子或原子团,它们有很高的化学活性。自由基中的未成对电子用黑点“·”表示。例如原子氢H·、氢氧基HO·、甲基CH3·、乙酰基CH3CO·等。自由基是很不稳定的,它不能长期存在,很容易继续发生反应。某些稳定分子中也有不成对电子,如O2、NO、NO2等,但不称为“自由基”。
[催化剂]能改变化学反应速度而在反应前后本身的化学组成、化学性质及数量没有变化的物质称为催化剂。正催化剂能加快反应速度,负催化剂(阻化剂)能减慢反应速度。
[催化反应]有催化剂参加的反应。若催化剂和反应物同处于均匀的气相或液相中,称为均相催化。若催化剂和反应物不在同一相中,且反应仅在催化剂与反应物的界面上进行,则称为多相催化。还有一类催化反应叫生物催化,或称酶催化,它既不同于均相催化也不同于多相催化,而是兼有二者的某些特征。
[阻化剂]见催化剂条。
[均相催化]见催化反应条。
[多相催化]见催化反应条。
[载体]工业上常常将催化剂附着在一些多孔性物质上作为催化剂的骨架,这种物质就称为催化剂的载体。载体主要有以下的作用:
⑴将催化剂附着在载体上可使催化剂有较大的表面,从而提高其催化活性。
⑵附着在载体上的催化剂只是很薄的一层,因此可大大节省催化剂的用量,这对贵金属催化剂特别重要;
⑶增强了催化剂的机械强度。
⑷选择导热性较好的载体还可有助于催化剂散热,能延长催化剂的使用寿命。常用的载体有浮石、硅胶、硅藻土、活性炭等。
[助催化剂]本身没有催化作用或只有很小的催化活性,但将它和某种催化剂组合后能显著提高催化剂的活性、选择性并延长催化剂的使用寿命,这种物质叫助催化剂。例如合成氨所用的Fe催化剂中,加入少量的助催化剂K2O和Al2O3可使Fe的催化效率明显提高。
[自催化作用]不需外加催化剂,而由反应生成物起催化剂的作用,叫自催化作用。例如酸性高锰酸钾和草酸的反应中,反应生成物Mn2+离子就能起到该反应的催化作用:
2MnO4-+5C2O42-+16H+=2Mn2++10CO2↑+8H2O
这类反应开始时速度较慢,随后逐渐加快。
[催化剂的选择性]催化剂的选择性有两方面的含义:
⑴不同类型的反应需要选择不同的催化剂;即便是同一类型的反应,使用的催化剂也不一定相同,如SO2的氧化用的是V2O5催化剂,而乙烯氧化却用金属Ag催化剂。
⑵对相同的反应物,如果选用不同的催化剂,可以得到不同的产物,例如乙醇的分解:
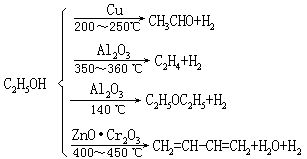
从热力学观点分析,以上的反应都能自发地进行,某种催化剂只对某一特定反应有催化作用,并不能加速所有可能发生的反应。
[催化剂中毒]反应体系中含有少量杂质就能严重降低甚至完全破坏催化剂的活性,这种物质称为催化剂毒物,这种现象称为催化剂中毒。催化剂中毒现象有两类:
⑴暂时性中毒(或称可逆性毒化),如合成氨生产中的O2、H2O(气)、CO、CO2等杂质引起的中毒。只要不断用纯净的原料气吹过中毒的催化剂表面,就可使毒物除去。
⑵永久性中毒(或称不可逆毒化),如合成氨生产中的含硫化合物及PH3等属于永久毒物。这主要是因为这类毒物与催化剂表面形成了牢固的表面化合物。
[体系]作为研究对象的一定物质或空间所组成的整体,也称系统。体系以外的其他物质或空间则称作环境。例如研究硝酸银和氯化钠在水溶液中的反应,含有这两种物质的水溶液就是体系,而盛溶液的烧杯、溶液上方的空气等就是环境。热力学体系可分三种:孤立体系、封闭体系、敞开体系。
[环境]指所研究的物质体系以外的其它部分(见体系条)。关于生态环境详见“环境部分”。
[敞开体系]体系与环境之间既有物质交换,又有能量交换。
[封闭体系]体系与环境之间没有物质交换,只有能量交换。
[孤立体系]体系与环境之间既没有物质交换,又没有能量交换。
[状态]即体系的状态,在热力学中用体系的性质来规定其状态。决定体系状态的性质有温度、压力、体积、组成等,比如当研究的对象是一定量的纯净气体时,温度和压力一定时,体系的状态就定了。
[状态函数]用于规定体系的热力学状态的宏观性质,如体积、温度、压力、物质的量等都叫做状态函数。状态函数的变化只取决于体系的始态和终态,而与变化的途径无关。
[压力]物理学中压强的概念,在化学热力学中常称作压力。单位是帕斯卡(Pa),压力为1Pa的含义是:1平方米面积上受到的垂直作用力为1牛顿(1Pa=1Nm-2)。
[压强]见压力条。
[理想气体]忽略了分子本身的体积和分子间作用力的气体。这种气体是不存在的,这只是一种理想状态,当真实气体处在较高的温度和较低的压力的状态下可近似地看成是理想气体。
[理想气体状态方程]即PV=nRT。式中P为气体压力,V为气体体积,n为气体的摩尔数,R为气体常数,T为热力学温度。
[气态方程]见理想气体状态方程条。
[气体常数]理想气体状态方程中的一个常数,常用R表示,R=8.314J·mol-1·K-1。
[广度性质]也叫容量性质,是体系宏观性质的一类。广度性质的数值与体系中物质的数量成正比,例如体积、质量、内能等。
[强度性质]体系的宏观性质的另一类,其数值与体系中物质的数量无关,仅由体系本身的性质决定,没有加和性,整个体系的强度性质与体系各个部分的强度性质的数值都相同。例如温度、密度、比热、气体压强等。
[等温过程]在保持温度不变的情况下(指体系的始态温度与终态温度相同),体系所进行的各种化学或物理的过程。在这种过程中体系和环境间可能有热和功的交换。
[等压过程]在恒定压力下(指体系的始态压力与终态压力相同,并等于环境的压力),体系所进行的各种化学或物理的过程。在这种过程中体系和环境可能有热量和功的交换。
[等容过程]在保持体积不变的情况下,体系所进行的各种化学或物理的过程。在这种过程中,体系和环境间,可能有热量的交换。
[绝热过程]在体系跟环境间没有热量交换的情况下,体系所进行的各种化学或物理过程。例如在有良好的绝热壁的容器内发生的化学反应,可认为是个绝热过程。另外,某些过程进行极迅速,来不及和环境进行热交换,如气体向真空膨胀,也可视为绝热过程。
[热力学可逆过程]对体系发生变化时所进行的过程,能通过原来过程的反方向变化而使体系回复到原来状态,同时在环境中没有留下任何永久性变化的过程,称为热力学可逆过程。在这过程进行中的每一瞬间,体系和环境都非常接近于平衡态。在等温的可逆过程中,体系对环境所作的功为最大功,环境对体系所做的功为最小功。可逆过程是一种理想的极限过程。有些实际过程,例如液体在气-液平衡下等温蒸发;原电池在外加电压接近或等于原电池电动势的情况下放电或充电的过程等,都可以近似看成可逆过程。
[热力学温标]又称绝对温标或开氏温标,是最基本的温度标定方法。热力学温度用符号T表示,其单位是K(Kelvin的缩写),是国际单位制(SI)的基本单位之一。热力学温标的1度是水的三相点热力学温度的1/273.16。
[绝对温标]见热力学温标条。
[开氏温标]见热力学温标条。
[摄氏温标]规定在101325帕斯卡(1大气压)下水的冰点为0度,沸点为100度,中间分100等分,每等分代表1度。摄氏温标的符号为t,单位是摄氏度用℃表示,水的三相点温度为0.01℃。
[焦耳]是能量(功、热)的单位,是SI的导出单位,用“J”表示,1J=1N·m(1焦耳=1牛顿·米)。
[内能]体系的内能就是体系内部所包含的一切能量,它包括体系内分子运动的动能、分子间相互作用的位能,以及分子内各种粒子(原子、电子、原子核)所具有的能量。但体系本身的动能和位能不包括在内。内能是体系本身的性质,仅决定于体系的状态,在一定的状态下,体系的内能应有一定值,内能是体系的状态函数。内能的绝对值现在还无法测量,然而对热力学来说,重要的不是内能的绝对值而是内能的变化值,这是可以用实验测量的。
[焓]体系的状态函数,用符号H表示。焓的定义式是:H=U+pV。U是体系的内能,p是体系压力,V是体积。在一定状态下,体系的焓应有一定值,但现在无法测定焓的绝对值,对热力学来说重要的是焓的变化值,这是可以通过实验测量的。
[焓变]体系终态的焓(H2)与始态的焓(H1)的改变量,用△H表示,△H=H2-H1。
[热效应]体系在一定温度下(等温过程)发生物理或化学变化时(在变化过程中只做膨胀功而不做其它功),所放出或吸收的热量。化学反应中的热效应又称反应热。根据化学热力学的分析,恒压下的反应热(又称恒压反应热)等于体系的焓变:△H=Qp,△H=(∑H)产物-(∑H)反应物。
[放热反应]放出热量的化学反应叫放热反应。在等温等压条件下,放热反应的焓变为负值,△H<0。
[吸热反应]吸收热量的化学反应叫吸热反应。在等温等压条件下,吸热反应的焓变为正值,△H>0。
[反应热]见热效应条。
[中和热]在稀溶液中酸碱中和生成1摩尔水的反应热。一元强酸与强碱的中和热约为57千焦,与酸碱种类无关,因为这实际上是1摩尔H+与1摩尔OH-反应生成1摩尔H2O的反应热。弱酸、弱碱以及多元酸碱的中和热,因有电离热的影响,不是定值。
[生成热]在热力学标准态下,由稳定单质生成1摩尔化合物时的反应热,叫标准生成热,简称生成热。根据热力学规定,在所有温度下稳定单质的生成热为零,这样化合物的生成热就可通过实验测定。
[燃烧热]指1摩尔纯物质完全燃烧,生成稳定的氧化物时的反应热。
[盖斯定律]1840年,盖斯(Гecc)在大量实验的基础上提出:“在等容或等压条件下,一个化学反应不管是一步完成还是分几步完成,其热效应总是相同的。”也就是说,在等容或等压条件下,反应热只与反应的始态和终态有关,而为反应的途径无关。盖斯定律的提出奠定了热化学的基础,它的重要意义在于可根据已经准确测定的反应热来计算难于测量的反应热的热效应。
[升华热]由1摩尔固态物质生成气态分子或原子所需要的热量。如1摩尔固态金属钠升华成气态钠原子,升华热为108.37kJ·mol-1。
[气化热]在温度不变的条件下,单位质量的液体转化为气体时所吸收的热量。如100℃(373.15K)时水的气化热为2253千焦/千克(539千卡/千克)。当在温度不变的条件下,1摩尔液体转化为气体时所吸收的热量,则是该液体的摩尔气化热,如100℃(373.15K)时,水的摩尔气化热为40.55千焦/摩尔(9.702千卡/摩尔)。
[溶解热]指1摩尔物质在25℃(298K),101325Pa(1大气压)下形成无限稀的溶液(溶液稀到再加溶剂时无热效应)时的热效应。常用∞aq表示大量水或无限稀的溶液。1摩尔HCl气体的溶解热为:
HCl(g)+∞aq=HCl·∞aq
△H=-75.4千焦
[有用功]指非膨胀功。体系在发生变化的过程中,除膨胀功以外的其它功叫有用功。例如电池在恒温恒压下放电,所做的电功即有用功,而电池中的物质由于化学反应而引起体积变化所做的功叫膨胀功。
[熵]体系的状态函数,是体系内部质点混乱程度(或无序度)的量度,常用S表示。当体系内质点的聚集状态发生改变时,其熵值就会改变,体系终态熵值(SB)与体系始态熵值(SA)之差为体系的熵变,用△S表示,△S=SB-SA。当体系的状态改变时,体系的熵变等于由始态到终态的任一可逆过程的热温商之和,其数学表达式为:
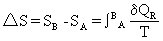
化学反应的熵变等于生成物的熵值与反应物的熵值之差,△S=(ΣS)生成物-(ΣS)反应物。
[熵变]见熵条。
[熵增大原理]见热力学第二定律(3)条。
[自由能]体系的状态函数,常用符号G表示,G=H-TS,自由能也称自由焓或吉氏函数。体系的自由能改变△G,为体系终态的自由能与始态的自由能之差。在等温等压条件下,体系的自由能减少等于体系所做的最大有用功。对等温等压条件下发生的化学反应,可以根据反应的自由能变[△G=(ΣG)产物-(ΣG)反应物],判断反应能否自发进行:当△G<0,反应发生时会放出自由能,可被利用来对环境做有用功,这个反应就能自发进行;当△G>0,必须由环境提供有用功反应才能发生,这个反应就是非自发的;当△G=0,则反应体系处于平衡状态。
[自由焓]见自由能条。
[吉氏函数]见自由能条。
[功函]体系的状态函数,常用符号F表示,F=U-TS,功函又称赫氏函数。在等温等容的条件下,体系功函的减少等于体系所做的最大有用功。
[赫氏函数]见功函条。
[热力学第零定律]两个热力学体系中每一个都和第三个热力学体系处于热平衡,那么它们彼此也必然处于热平衡。这就是热力学第零定律。
[热力学第一定律]即能量守恒和转换定律。可以这样表述:
⑴不供给能量而可连续不断产生能量的机器叫第一类永动机,第一类永动机是不可能存在的。
⑵在体系状态变化过程中,它的内能改变等于在这个过程中所做的功和所传递的热量的总和。
当某体系的状态改变时,假设体系吸收的热量为Q焦耳,同时对环境做了W焦耳的功,根据热力学第一定律,应有下面的公式:△U=Q-W。
[热力学第二定律]是热力学的基本定律之一,有多种表述方式:
⑴热量总是从高温物体(体系)传到低温物体,不能自发地进行相反的传递。
⑵功可以全部转化为热,但任何热机不能全部地、连续地把所获得的热量转变为功。
⑶在任何自发过程中,体系和环境的总熵值是增加的。
热力学第二定律所要解决的中心问题是自发过程的方向和限度。
[热力学第三定律]当体系的热力学温度趋于零时,混乱度为最小,此时体系的熵值也趋于零。也可以说,在绝对零度时,任何纯物质的完美晶体的熵值都等于零。有了热力学第三定律,从原理上说,纯物质的绝对熵值的求算问题就可以认为是解决了。
[分压]即分压力。恒温时混和气体中某组分气体单独占有与混和气相同的体积时,对容器所产生的压力。混和气的总压就等于各组分气体的分压之和。
[道尔顿分压定律]1807年由道尔顿(Dalton)首先提出。内容是:混和气体的总压力等于各组分气体的分压力之和。由此可进一步得出:某组分气体分压的大小和它在混和气中的体积分数(或摩尔分数)成正比,等于混和气体的总压乘以该组分的摩尔分数。这个定律的数学表达式为:
P总=P1+P2+P3+…+Pi
Pi=p总Xi
(Pi为某组分的分压,Xi为该组分的摩尔分数)。
[物态]当大量的微观粒子在一定的温度和压力下相互集聚为一种稳定的结构状态时,叫做物质的一种状态,简称物态。若只从物体的宏观特征看,物质有三态,即固态、液态、气态。
[相]体系中具有相同组成、物理性质和化学性质完全均匀的部分。相与相之间存在着界面,越过此界面,性质就有一个突变。不仅限于固、液、气三相之间。两种不同结晶的晶体,虽都是固体,也是两个相。
[相律]在平衡体系中,联系体系内相数(Φ)、组分数(K)、自由度数(f)及影响物质性质的外界因素(如温度、压力、重力场、磁场、表面能等)之间关系的规律。若只考虑温度和压力,而不考虑其它因素的影响,平衡体系中相数、组分数和自由度之间的关系为:
f=K-Φ+2
由上式可以看出,体系每增加一个组分数,则体系的自由度数就增加一个;如果体系增加了一个相数,则自由度就要减少一个。
[自由度]欲表明一体系在某状态时的强度性质所必需的独立变量数,称为该体系的自由度。例如,要表明一定量水所处的状态,需要指定水所处的温度和压力;若只指定温度时,则水的状态还不能确定,当指定了温度和压力,则水的状态就确定了,不能再任意指定其它性质(如密度等),由此可知,当体系中只有水存在时,体系的自由度等于2。在一定范围内,同时改变温度和压力这两个因素,仍能保持水的液相而不产生新相。
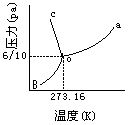
[相图]当体系中有多相存在时,把体系的相平衡规律用几何图形展现出来就成为相图。它能直观地表明体系的状态与温度、压力、组成的关系。
下图为水的相图。图中OA线是液-气平衡线(即水的蒸气压曲线),OB线是固-汽平衡线(即冰的蒸气压曲线),OC线是固-液平衡线,O点是冰-水-气三相平衡的三相点。此单组分体系相图上,有三个单相面,三条两相平衡线,一个三相点,与相律完全一致。
[相变]在某一体系中,各相之间的相互转变称作相变。例如,物质的气、液、固三态间的转变;固态物质的不同晶型间转变等。
[相平衡]指在某一体系中,由两个相或多个相形成的平衡状态。
[均相体系]又称单相体系,即只有一个相的体系。如二氧化氮NO2和四氧化二氮N2O4两种气体共存的体系。
[单相体系]见均相体系条。
[均相反应]参加化学反应的物质都在同一相中,这种反应叫均相反应。此类反应又称单相反应。
[均相平衡]又称单相平衡。指某一体系中由一个相组成的平衡状态,就是在一个相-气相组成的平衡。例如,合成氨的反应:N2(气)+3H2(气)2NH3(气)
[单相平衡]见均相平衡条。
[多相体系]又称非均相系,指含有两个或多个相的体系。
[非均相体系]见多相体系条。
[多相反应]参加化学反应的物质不是在同一相中,这种反应叫多相反应。例如,在固体催化剂表面进行的反应;金属固体在酸溶液中发生的反应等。此类反应又称非均相反应。
[多相平衡]又称非均相平衡。某一体系中由两个或多个相组成的平衡状态。例如,由冰、水、水蒸气形成的多相物理平衡;又如在封闭容器中碳酸钙受热分解可形成多相物理平衡:
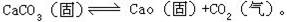
[三相点]指单组分体系中三相(一般指气、液、固三相)处于平衡共存时的温度和压力。在单组分体系中最多只能三相共存,这在相图中为一个点。例如,固相的冰、液相的水和气相的水蒸气三相平衡共存在温度为0.01℃(273.16K)、压力为610帕(4.6mmHg),这就是水的三相点(见图1)。
[恒沸混和物]又称共沸混和物。若某二组分溶液的组成与其蒸气的组成相同,在此浓度的溶液用分馏方法分离出纯组分是不可能的。这种溶液与一般溶液不同,由开始沸腾到蒸发终了,蒸气的组分与溶液的组成始终相同,其沸点不变。这种溶液称为恒沸混和物。
[共沸混和物]见恒沸混和物条。
[最低恒沸点]当恒沸混和物的沸点低于任一纯组分的沸点时,称为“最低恒沸点”。
[最高恒沸点]当恒沸混和物的沸点高于任一纯组分的沸点时,称为“最高恒沸点”。
[低共熔物]又称低共熔混和物。两种或两种以上的物质组成的熔体,当其组成达到一定值时,可在一最低凝固温度,以比较均匀的形式共同析出固体,该固体若加热,仍在此最低温度共同熔融成熔体。该固体称低共熔物,此温度称低共熔点。
[低共熔混和物]见低共熔物条。
[低共熔点]见底共熔物条。
[饱和蒸气压]在一定温度下,在密闭容器中,当液体的蒸发速度与蒸气的凝聚速度相等时,液体和它的蒸气处于平衡状态。该液态平衡的蒸气称为饱和蒸气,饱和蒸气所产生的压力称为饱和蒸气压。饱和蒸气压简称蒸气压,是所有液体和某些固体的特有性质。
[蒸气压]见饱和蒸气压条。
[蒸气压降低]在一定温度下,溶液的蒸气压总是低于纯溶剂的蒸气压。对难挥发非电解质的稀溶液,蒸气压降低的数值只与溶液中溶质的摩尔分数有关,而与溶质的本性无关,溶液浓度越大,蒸气压下降的数值就越大。由于溶液蒸气压降低导致溶液的沸点高于纯溶剂的沸点。
[沸点升高]溶液蒸气压的下降导致了溶液沸点的升高。溶液沸点升高的程度只与溶液的浓度有关,难挥发非电解质稀溶液的沸点升高数值和溶液的质量摩尔浓度成正比,可用公式表示:△Tb=Kbm。式中△Tb是沸点上升的度数,Kb是溶剂的沸点上升常数,m是溶液的质量摩尔浓度。
[凝固点]晶体物质凝固时的温度,即该物质的液态和固态平衡共存时的温度。凝固点决定于晶体的种类和所受的压强。在一定压力下,任何晶体的凝固点和它的熔点相同。而非晶体则无一定的凝固点。
[冰点]即水的凝固点。冰点与压力的大小有关,在101325帕斯卡(1大气压)的压力下,冰点为273.15K。水的冰点随外压增大而降低。
[凝固点降低]溶液的凝固点低于纯溶剂的凝固点的性质。溶液凝固点降低的程度只与溶液的浓度有关,而与溶质的本性无关。可用公式表示:△Tf=Kfm。式中△Tf是凝固点降低的度数,Kf是溶剂的凝固点降低常数,m是溶液的质量摩尔浓度。
[渗透压]当溶液与纯溶剂(或两种浓度不同的溶液)用半透膜隔开时,溶剂分子能通过半透膜从纯溶剂一边进入溶液,或从稀溶液一边进入浓溶液,这种现象叫渗透现象。在溶液与纯溶剂组成的体系中,欲阻止渗透现象发生,必须增大溶液的压力,刚刚足以阻止渗透过程进行所外加的压力叫渗透压。对于非电解质稀溶液来说,溶液的渗透压与溶液的浓度成正比,而与溶质的本性无关,渗透压的计算公式为:π=RTC。式中π为溶液渗透压,R为气体常数,T为绝对温度,C为溶液的摩尔浓度。
[拉乌尔定律]1887年由拉乌尔(Roult)提出。内容是:在一定温度下,稀溶液中溶剂的饱和蒸气压等于纯溶剂的饱和蒸气压乘以溶剂的摩尔分数。可用公式表示:
pA=p0AxA
式中pA代表稀溶液中溶剂的饱和蒸气压,p0A代表纯溶剂的饱和蒸气压,xA代表溶液中溶剂的摩尔分数。
[亨利定律]1803年由亨利(Henry)指出。内容是:在一定温度下,当液面上的一种气体与溶液中所溶解的该气体达到平衡时,气体的平衡分压与被溶解的气体在溶液中的浓度成正比。可用公式表示:
p=Kxx
式中p为所溶解的气体在溶液液面上的平衡分压,Kx为比例常数,x为溶液中溶解的气体的摩尔分数。如果气体在溶液中的浓度以质量摩尔浓度(m)或体积摩尔浓度(c)表示,则亨利定律相应的数学表达式为:
p=Kmm
p=KCC
应注意以上三个式子中Kx≠Km≠Kc。亨利定律适用于稀溶液和气体分压不太大,且溶质在气相中和溶液中分子状态相同的情况。
[摩尔热容]使1摩尔物质温度升高1度所需要的热量,是该物质的摩尔热容。摩尔热容的单位是焦耳每摩尔开尔文,记作焦/摩·开,国际通用符号是J·mol-1·K-1。
[热力学标准态]化学热力学中,为计算各种物系的热力学函数而规定的特定状态,简称标准状态。指在一定温度下[一般是25℃(298.15K)],纯净物质处于101325帕斯卡(1大气压)时,若是多组分体系,所指定的组分活度应等于1,气体物质的分压应等于101325帕斯卡(1大气压)时的状态。
[标准状态]见热力学标准态条。
[化学平衡]可逆反应中正、逆反应速度相等,在温度和压力不变的情况下,反应物和生成物的浓度不再随时间而改态的状态。化学平衡是动态平衡,它受温度、压力和浓度的影响。
[化学平衡常数]系在一定温度下,可逆反应达到化学平衡状态时,生成物浓度的反应系数次幂的乘积与反应物浓度的反应系数次幂的乘积之比值,这个比值在一定温度下是个定值。例如对可逆反应:
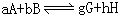
用浓度表示的平衡常数为:
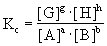
式中[A]、[B]、[C]、[D]代表平衡体系中各组分的浓度。
也可用各组分的摩尔分数计算平衡常数,以Kx表示(X代表组分的摩尔分数):
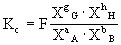
对于有气体参加和生成的反应,也可用各组分的分压计算平衡常数,以Kp表示:
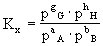
以上三种平衡常数可以进行如下换算:
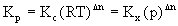
式中R为气体常数、T为绝对温度、p为体系总压,△n=(g+h)-(a+b)。
[勒沙特列原理]又称平衡移动原理。1887年由法国化学家勒沙特列(Le chatelier)提出。其内容是:如果改变影响平衡的一个条件(如浓度、压力、温度等),平衡就向能够减弱这种改变的方向移动。比如,在温度、压力不变的条件下增加反应物的浓度或减少生成物的浓度,平衡朝正反应方向移动;在温度不变的情况下,对反应前后分子数有变化的气体反应,增大压力,平衡朝分子数减少的方向移动;在其它条件不变时,升高温度,平衡就朝吸热反应方向移动。勒沙特列原理在生产上有广泛的应用,利用这一原理选择适当的反应条件,可以提高产率、降低成本。
[范特荷甫等温方程]又称化学反应等温方程式。由荷兰物理化学家范特荷甫(Van′t Hoff)提出,假设有一理想气体的化学反应是:

此体系的自由能变为:
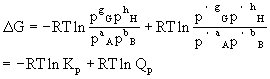
式中P代表该反应达到化学平衡状态时体系中各组分的分压,Kp代表平衡常数;而p′代表任意状态时反应体系中各组分的分压,Qp不是平衡常数,为区别起见,称Qp为“分压商”。用范特荷甫等温方程式可以判断在一定条件下,某化学反应进行的方向和限度。在等温等压除膨胀功不做其它功的情况下:
当Qp<Kp时,△G<0,正反应能自发进行。
当Qp>Kp时,△G>0,正反应不能自发进行,而逆反应能自发进行。
当Qp=Kp时,△G=0,反应达到平衡状态。
若反应体系中的气体不遵守理想气体规律,则应当用逸度(f)来代替压力,则化学反应等温方程式为:
△G=-RTlnKf+RTlnQf
如果反应是在稀溶液中进行,则化学反应等温方程式为:
△G=-RTlnKc+RTlnQc
式中Kc为浓度平衡常数,Qc为“浓度商”。
对在非理想溶液中进行的化学反应,应以活度(a)来代替浓度,即:
△G=-RTlnKa+RTlnQa
[玻恩-哈伯循环法]由玻恩(Born)和哈伯(Haber)提出的求某一过程能量变化的方法。此法建立在热力学第一定律的基础上,根据某过程总的能量变化等于各个分过程能量变化的总和的原理,把某个变化过程设计成由若干个分过成组成的热力学循环,即玻恩-哈伯循环。这样,就可以根据已知的一些能量变化求出其中某一过程的能量变化。以求NaCl的晶格能为例,设计出生成氯化钠晶体的热力学循环:
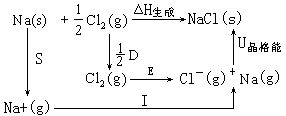
其中各个过程的能量变化是:△H生成=411.0kJ·mol-1,为NaCl晶体的生成热;S=108.8kJ·mol-1为晶体钠的升华热;I=493.29kJ·mol-1为钠原子的第一电离能;1/2D=119.7Kj·mol-1为1/2 mol氯气的解离能;E=-361.9kJ·mol-1为氯原子的电子亲和能;U晶格能为NaCl的晶格能是未知量。根据热力学第一定律可得出关系式:
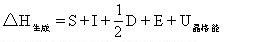
经过变形可求出NaCl的晶格能:
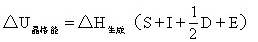
=-411.0-(108.8+493.29+119.7-361.9)
=-770.9kJ·mol-1
[晶格能]在298.15K和101325Pa(1大气压)下,由相互分离的气态阳离子和气态阴离子,生成1摩尔离子晶体时所释放的能量。晶格能可以衡量离子晶体中离子键的强弱和晶体的热稳定性。晶格能可以通过热化学实验间接测定,还可以通过热化学循环计算(见玻恩-哈伯循环法)以及根据静电吸引理论计算。
[转化率]某反应体系达到化学平衡状态时,其中某个指定反应物的转化率是该物质在达到平衡时已转化的量占反应起始时该物质总量的百分率。可用下式进行计算:

(式中“量”可用质量,也可用物质的量。)
[原子]由希腊词义“不可分”衍化来的。原子是元素能够存在的最小单位,是化学变化中的最小微粒。原子是由原子核和核外电子组成的。原子核由质子和中子组成。在化学反应中,只是核外电子发生了变化,原子核的组成并没有改变。在核反应中,原子核发生了变化,原子变成了其它元素的原子。
[原子核]由质子和中子组成的带正电荷的微粒。原子核所带的正电荷数(核电荷数)等于核内质子数。原子的质量主要集中在原子核上,原子核的体积只占原子体积的几千亿分之一。原子核内的质子和中子由结合能结合在一起。
[质子]原子核的组成部分,是原子核中带正电荷的基本粒子。元素的原子序数就是核内的质子数。质子的质量是1.67265×10-27千克,即1.00728原子质量单位。
[中子]原子核中不带电的基本粒子。质量是1.67495×10-27千克,即1.008665原子质量单位。除11H外,每种核素的原子核都含有中子。
[电子]原子中带负电的微粒。质量是9.11×10-31千克,半径是2.82×10-15米。原子由原子核和核外电子组成,核外电子数与质子数相等。原子中的电子受到外来能量的激发能脱离原子核的束缚,以电流形式通过导体、电子管或半导体。放射性核如镭能以β-粒子的形式放出电子。
[基本粒子]泛指比原子核还小的物质单元。包括光子、电子、质子、中子以及在宇宙射线和高能物理实验中发现的一系列粒子。现已发现的基本粒子有几百种,并分为轻子(如电子、正电子、中微子等)和强子(如质子、中子、介子等)两大类。许多基本粒子都有对应的反粒子。
[反粒子]许多基本粒子如电子、质子、中子、中微子、介子、超子等,都有对应的正电子、反质子、反中子、反中微子、反介子、反超子等,这些对应的粒子总称为反粒子。
[亚原子粒子]指小于原子的粒子,即基本粒子或原子核。
[原子实]原子中,原子核及除价电子以外的内层电子组成原子实。例如,钠Na的核外电子排布为:1s22s22p63s1,其中第1、2电子层与原子核组成原子实,此原子实与氖Ne(1s22s22p6)的结构相同,钠的核外电子排布也可写作[Ne]3s1。钾K的电子排布为:1s22s22p63s23p64s1,也可写作[Ar]4s1。铜Cu为:1s22s22p63s23p63d104s1,也可写作[Ar]3d104s1。
[原子半径]通常指以实验方法测定的相邻两同种原子的核间距离的一半。原子的大小无严格边界,无法精确测定一个单独原子的半径。目前所使用的原子半径数据只有相对的、近似的意义。根据测定的方法不同,有三种原子半径数据:
⑴共价半径:同种元素的两原子以共价键结合时,两核间距离的一半,称该元素原子的共价半径。
⑵金属半径:金属晶体中,相邻两原子核间距离的一半,称该元素原子的金属半径。
⑶范德华半径(简称范氏半径):在分子型晶体中,不属于同一分子的两个最接近的相同原子核间距离的一半,称该元素原子的范氏半径。
一般说来,各元素的金属半径比共价半径略大,而范德华半径则比共价半径大得多。
[氢光谱]指氢原子的线状光谱。装有氢气的放电管,通过高压电流,氢分子分解为原子并受到激发,激发态氢原子中的电子,由高能级跃迁回低能级时,以发光形式释放能量。这种光通过棱镜时,可以看到几条分立的波长不同的谱线,即氢光谱。1913年丹麦物理学家玻尔(Bohr)首先用量子概念解释了这一现象。
[玻尔理论]由丹麦物理学家玻尔在1913年提出的阐明原子结构的初步理论。它的基础是两个基本假设:
⑴核外电子沿一定轨道绕核运转,在稳定轨道上运动的电子不放出能量,也不吸收能量;
⑵在一定轨道上运动的电子都具有一定的能量,这些能量具有量子化的特征,只能取某些由量子化条件决定的分立数值。这些不连续的能量状态称为电子的能级。
玻尔理论冲破了经典物理学中能量连续变化的束缚,大胆地把量子概念引进原子结构理论,解释了用经典物理学无法解释的氢原子结构与氢光谱的关系。但玻尔理论未能认识电子运动的另一重要特征--玻粒二象性,未能完全摆脱经典物理学的束缚,以致认为核外电子运动也象宏观物体那样具有固定的轨道,在解释多电子原子的光谱时,遇到了难以解决的困难。这些问题直到量子力学建立后,才得到解决。
[玻尔轨道]玻尔理论中电子绕核运行的许多分立的轨道。在一定轨道上运动的电子都具有一定的能量,也称作能级。电子从外层轨道向内层轨道跃迁时,以发光形式释放能量,其能量变化具有量子化的特征。
[玻尔半径]在玻尔理论中,它是氢原子处于基态时,电子沿圆形轨道绕核运行的半径,其值为5.29177×10-11米,常用a0表示。在原子物理学中,a0也是一种长度单位。
[基态]微观粒子系统(如原子、原子核、或其它多粒子体系等)所具有的各种状态中能量最低的状态。此种状态为最稳定的状态。
[激发态]微观粒子系统当其内部能量高于基态能量时所处的运动状态。原子获得能量(如吸收光子)可处于激发态。激发态原子是不稳定的,一般将通过发射光子或与其它粒子发生作用而回复到基态。
[测不准原理]由德国物理学家海森堡(Heisenberg)提出。他认为微观粒子的某些成对的物理量不可能同时具有确定的数值,如位置和动量之间有以下测不准关系:
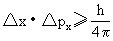
式中△x是确定位置的测不准量,△px是确定动量的测不准量,h是普朗克常数。上式表明,微观粒子位置测定得愈准确(△x愈小),则动量测定的准确程度就愈差(△px愈大),两者乘积大于或等于h/4π,反之亦然。时间和能量也服从测不准关系,它的意义是微观粒子存在于某一能量状态的时间愈短,则这一能量的确定程度愈差。
[测不准关系]见测不准原理条。
[波粒二象性]微观粒子的基本属性之一。它们有时显示出波动性(这时微粒性不显著),有时又显示出微粒性(这时波动性不显著),这种在不同条件下分别表现为波动性和微粒性的特征称为波粒二象性。1905年爱因斯坦(Einstein)提出的光子学说阐明了光的这一特征。1924年德布罗意(deBroglie)提出电子和其它实物粒子也具有波粒二象性的假说,这在1927年被实验证实。从此,在科学上就认为一切微观粒子都具有波粒二象性。粒子的质量或能量愈大,波动性愈不显著。所以,可认为宏观物体不具有波动性。
[德布罗意波]由法国物理学家德布罗意(Louis Vi-ctor de Broglie)在1924年首先提出。他把当时已发现的光具有波粒二象性这一事实加以推广,预言一切微观粒子都具有波粒二象性,并提出著名的德布罗意公式:
λ=h/mv
式中λ为与质量为m,运动速度为v的实物微粒相应的波长。德布罗意波又称物质波。
[德布罗意公式]见德布罗意波条。
[量子]微观世界的某些物理量不能连续变化,只能以某一最小单位的整数倍发生变化,这个最小单位就称为该物理量的量子。例如,物质吸收或发射的能量只能是某一最小能量单位的整数倍,其能量量子的大小为hv(v为幅射频率,h为普朗克常数)。光的能量变化也具有量子化的特征,光的能量量子(光子或光量子)的大小也是hv。
[量子化]见量子条。
[量子论]量子力学的先驱,又称旧量子论。从普朗克在物理学中提出量子概念(1900年)开始,特别是在玻尔提出氢原子结构理论(1913年)后发展起来的。它在解释微观粒子运动中,引进了运动具有量子特征的条件,但未能完全摆脱经典物理学的束缚。它是探索微观粒子运动的初步理论,它的进一步发展导致量子力学的建立。
[量子力学]研究微观粒子(电子、质子、中子、原子、分子等)运动规律的理论,是现代物理学的基础理论之一。它是法国物理学家德布罗意、奥地利物理学家薛定谔(Erwin Schr dinger)、德国物理学家海森堡等人逐步建立和发展起来的。薛定谔方程是量子力学中描述微观粒子运动规律的基本方程,量子力学的建立对科学的发展有重要意义,它标志着人类对物质的认识从宏观世界进入到微观世界。
dinger)、德国物理学家海森堡等人逐步建立和发展起来的。薛定谔方程是量子力学中描述微观粒子运动规律的基本方程,量子力学的建立对科学的发展有重要意义,它标志着人类对物质的认识从宏观世界进入到微观世界。
[薛定谔方程]量子力学中研究微观粒子的基本方程,由奥地利物理学家薛定谔(Erwin Schrdinger)提出:
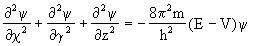
式中x、y、z为微观粒子(如电子等)的空间坐标,E是总能量,即电子的动能和势能之和,V为总势能,m是微观粒子的质量,h是普朗克常数,ψ是波函数。这一方程在量子力学中的地位就好象牛顿定律在经典力学中的地位同样重要。
[波函数]量子力学中表征微观粒子或其体系运动状态的函数。在原子中核外电子的运动状态就是用波函数ψ来描述的。ψ是薛定谔方程的解,是一个函数式。微观粒子(如电子)在空间某点出现的几率密度,跟波函数绝对值的平方│ψ│2成正比。
[原子轨道]在量子力学中是波函数的同义词。是指n、l、m(见主量子数、角量子数、磁量子数)具有一定值的波函数。每一个原子轨道都有一个相应的波函数。原子轨道又称原子轨道函数或原子轨函。
[原子轨函]见原子轨道条。
[几率]在数学上也称为“概率”、“或然率”。用来表示随机事件发生的可能性大小的一个量。必然发生的事件的几率为1,不可能发生的事件的几率为0,而一般随机事件发生的几率是在0与1之间(或0与100%之间)的一个数。因此,几率也可理解为“机会”的百分数。在现代原子结构理论中,常用来说明原子中核外电子在空间某处出现机会的多少。
[几率密度]指空间某处单位体积中出现的几率。在空间某一点,微观粒子(如电子等)出现的几率密度正比于波函数的绝对值的平方。在原子结构理论中,常用来说明原子中的电子在核外空间某处单位体积里出现机会的多少。
[电子云]是电子在核外空间出现几率密度分布的形象化描述法。也就是波函数的绝对值的平方│ψ│2在空间的分布的描述。电子云用小黑点表示,小黑点稠密的地方表示几率密度大,小黑点稀疏的地方表示几率密度小。电子云有时也用界面图表示,电子云的界面是一个等密度面,电子在界面以内的几率很大,而在界面以外的几率可以忽略不计。
[主量子数]表示电子在原子中运动状态的四个量子数之一,符号是n,是确定电子能量的主要量子数。它的取值是1、2、3、……等正整数,一般随着n值的增大,电子的能量随着增加,离核的平均距离也相应增大。在一个原子内,具有相同主量子数的电子,差不多在同样的空间范围内运动,所以,习惯上称主量子数相同的电子为一个电子层(或处于同一电子层)。常用K、L、M、N、O、P等符号分别表示n=1,2,3,4,5,6等电子层。
[电子层]见主量子数条。
[角量子数]表示电子在原子中运动状态的四个量子数之一,符号是l,是代表角动量的量子数,确定电子云的形状。当n值一定时,l只能取0,1,2,3……(n-1)等值。当l=0时,电子云的形状是球形对称的,用符号s表示,也称s亚层;当l=1时,电子云呈哑铃形,用符号p表示,也称p亚层;当l=2时,电子云常呈四瓣形,用符号d表示,也称d亚层;当l=3时,用符号f表示,也称f亚层,电子云形状更为复杂。在n=1的电子层中,l=0,只有一个亚层即1s亚层; n=2时, l=0,1,这时有两个亚层,即2s, 2p亚层;n=3时,l=0,1,2,这时有三个亚层,即3s,3p,3d。依此类推可知各电子层具有的亚层数目和种类。
[电子亚层]见角量子数条。
[磁量子数]表示电子在原子中运动状态的四个量子数之一,符号为m,确定电子云在空间的取向,是代表角动量在磁场方向的分量的量子数。当角量子数l值一定时,磁量子数m的取值为0,±1,±2,±3……±l,即有(2l+1)个数值。磁量子数有几个取值,就代表电子云在空间有几种取向,或者说有几个不同的轨道。例如,l=0时,m=0,表示s电子云无方向性,在空间只有一种取向,即只有一个轨道;l=1时,m=+1,0,-1,表示p电子云在空间有三种取向,即有三个轨道;l=2时,m=+2,+1.0,-1,-2,表示d电子云在空间有五种取向,即有五个轨道;余类推。
[等价轨道]又称简并轨道,指能量相等的原子轨道。例如n=2时的三个p轨道,2px,2py,2pz;其它如在主量子数相同时的五个d轨道,七个f轨道等,都是等价轨道。
[简并轨道]见等价轨道条。
[自旋量子数]表示电子在原子中运动状态的四个量子数之一,符号为ms,确定电子的自旋方向,是代表电子自身相反的两种运动的量子数。它仅有+1/2和-1/2两个取值。电子的两种相反自旋状态用箭头“↑”和“↓”表示。
[能级]微观粒子系统在定态中只能处于一系列不连续的、分立的能量状态。按这些状态的能量由低到高加以排列即组成能级。在原子体系中,能级的高低主要由主量子数决定,但在多电子原子中又会出现能级交错现象(见能级交错)。在分子轨道理论中,认为原子轨道组成分子轨道,分子轨道也按能量分为若干能级。
[屏蔽效应]在多电子原子中,将其它电子对某个指定电子的排斥作用归结为,核电荷吸引电子的作用受到抵消,这种效应叫屏蔽效应。这样,对指定电子来说,它所受到核的作用,可以看成是具有核电荷(Z-σ)的核的作用。Z是核电荷,σ是屏蔽常数,(Z-σ)是有效核电荷。一般说来,内层电子对外层电子的屏蔽效应较大,同一层电子之间屏蔽效应较小;外层电子对内层电子的屏蔽效应很小,可忽略不计。
[有效核电荷]见屏蔽效应条。
[钻穿效应]在多电子原子中,在核附近出现几率较大的电子,可以较好地避免其他电子的屏蔽作用,受到较大的有效核电荷的吸引,因而能量较低。这种现象称为钻穿效应。钻穿效应主要是由于电子出现的几率径向分布不同形成的。钻穿效应不仅能解释n相同,l不同时轨道能量的高低;而且可以解释当n和l都不同时,有的轨道发生的能级顺序交错的现象。
[能级交错]在多电子原子中,当原子处于基态时,主量子数较大的某些轨道的能量反而低于主量子数较小的某些轨道的能量的现象。例如,从第4周期开始,4s轨道能量反比3d轨道能量低,按能量最低原理,电子应先填充4s轨道,然后再填充3d轨道。产生能级交错的原因比较复杂,可能跟屏蔽效应、钻穿效应及其它影响轨道能量的因素有关。
[泡利不相容原理]1925年由奥地利物理学家泡利(Pauli)提出。原理指出,在同一原子中不可能有四个量子数完全相同的电子,即每个原子轨道最多只能容纳两个电子,并且这两个电子的自旋必须相反。从而可得出,第n层电子最多能容纳的电子数是2n2个。
[能量最低原理]在不违背泡利原理的情况下,核外电子总是尽先排布在能量最低的轨道上,只有当这些轨道占满后,电子才依次进入能量较高的轨道。在多电子原子中,电子能量的高低与主量子数n和角量子数l有关。当n≥3时,出现能级交错现象(见能级交错)。
[洪特规则]也称等价轨道原则。由德国物理学家洪特(Hund)提出。在同一亚层的各个轨道上排布的电子,总是尽先分占不同的轨道,并且自旋方向相同。这样,整个原子的能量最低。洪特规则也有特例,等价轨道在全充满(P6、d10、f14)、半充满(p3、d5、f7)、全空(p0、d0、f0)时,原子结构比较稳定。例如,24Cr的电子排布式是1s22s22p63s23p63d54s1;29Cu的电子排布式是1s22s22p63s23p63d104s1。
[电子排布式]表示原子核外电子排布的式子。可清楚地表明各亚层中的电子数目。例如17Cl的电子排布式是1s22s22p63s23p5。
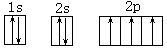
[轨道表示式]表示原子核外电子在各亚层、各轨道中排布的式子。能清楚地反映核外电子排布的规律。例如,7N的轨道表示式是:
[电离能]指从基态的气态原子、离子或分子,失去一个电子所需要的能量叫电离能,以千焦·摩尔-1为度量单位。元素的第一电离能指元素的基态的气态原子失去一个电子变为气态+1价阳离子所需要的能量。元素第一电离能的数值随原子序数的递增呈现周期性的变化。
[电子亲合能]指基态的气态原子、离子或分子得到一个电子所释放的能量。元素的电子亲合能指元素的气态原子得到一个电子变为气态-1价阴离子所释放的能量。从电子亲合能数据可以判断元素的原子得电子的难易。非金属元素一般具有比较大的电子亲合能。因此,非金属元素的原子比金属元素的原子容易得电子。
[电负性]分子中原子对成键电子吸引能力的相对大小的量度。目前有三套电负性数据:鲍林(Linus Pauling)电负性,用xp表示;密立根(Mulliken)电负性,用xM表示;阿莱-罗周(Allred-Rochow)电负性,用xA表示。三套电负性数据,是在不同的基础上计算出来的,因此它们的数值不完全相同,但都能反映元素的原子在化合物中吸引电子的能力。
[质能关系式]物质的质量m,与其能量E之间的关系式:E=mc2,式中c为真空中的光速。这是20世纪初爱因斯坦(Einstein)提出的狭义相对论的一个重要结论。由质能关系式可以看出,当物体的能量发生改变时,其质量也有相应的变化。运用这一关系可以解释原子核的质量亏损现象,认识到在原子核内蕴藏着巨大的能量,可以通过核反应加以利用。
[质量亏损]原子核的质量小于它所含各核子独自存在时的总质量,这两种质量的差额称为质量亏损。质量亏损说明,当核子集合组成原子核时要释放出结合能(即使核分解为质子和中子时所需要的能量)。它的数值越大,原子核就越稳定。
[离子]带电荷的原子或原子团。由原子、原子团或分子失去或得到电子时形成的。失去电子时形成带正电荷的阳离子(正离子),如H+、Na+、NH4+等;得到电子时形成带负电荷的阴离子(负离子),如Cl-、OH-、SO42-等。离子存在于离子化合物中,也存在于溶液中(和溶剂分子相结合),还存在于气体中。离子的性质与电中性的原子、原子团、分子的性质完全不同。
[阳离子]见离子条。
[正离子]见离子条。
[阴离子]见离子条。
[负离子]见离子条。
[离子半径]又称离子有效半径,是通过实验测定的。一般认为的离子半径是:离子晶体中相邻的两个正负离子的核间距是正负离子的半径之和。若核间距为d,则d=r++r-(r+,r-分别代表正负离子的半径)。已知一个离子的半径和核间距,就可以求出另一离子的半径。原子失去电子形成的阳离子的半径比其原子半径小,如Na原子半径为 1.537×10-10米,而Na+离子半径为0.95×10-10米。原子得到电子形成的阴离子的半径比其原子半径大,如Cl原子半径为0.99×10-10米,而Cl-离子的半径为1.81×10-10米。
[离子构型]指离子的外围电子构型。是离子的重要特征。离子构型主要可分为以下几种类型:
⑴惰气型,即与惰性元素原子的核外电子排布相同。其中有He型,如Li+(ls2)、Be2+(is2)等;Ne型,如F-(2s22p6)、Na+(2s22p6)、Mg2+(2s22p6)等;Ar型,如Cl-(3s23p6)、K+(3s23p6)等。
⑵18电子型,最外层为18个电子的离子,如Zn2+(3s23p63d10)、Ag+(4s24p64d10)等。
⑶18+2电子型,次外层为18个电子,最外层为2个电子的离子,如Sn2+(4s24p64d105s2)、Pb2+(5s25p65d106s2)等。
⑷不规则型,最外层有9-17个电子的离子,如Fe3+(3s23p63d5)、Cu2+(3s23p63d9)等。
[离子的极化作用]电荷相反的离子相互接近时就有可能发生极化,即它们在相反电荷的电场的影响下电子云发生变形。离子使异号离子极化而变形的作用称为该离子的极化作用。离子极化作用的大小跟离子所带电荷的多少、离子半径大小及离子构型有关。一般离子所带电荷数越大,半径越小,外层电子的屏蔽作用越小,离子的极化作用就越大。通常阳离子的极化作用较强,因此在多数情况下应考虑阳离子对阴离子的极化作用。有的阴离子对阳离子也有显著的极化作用,而阳离子被极化后又对阴离子产生更强的极化作用。这样反复作用叫做离子的相互极化作用或附加极化作用。离子的极化对化合物的键型、溶解度、熔点、颜色及化学稳定性都有一定的影响。
[离子的变形性]离子被异号离子极化而发生电子云变形的性能称为该离子的变形性或可极化性。离子变形性的大小主要和离子半径、离子的电子构型有关。最容易变形的是半径较大的阴离子和18电子外壳或不规则电子外壳的带电荷少的阳离子,如Ag+、Hg2+等。最不容易变形的是离子半径小,带电荷多的惰气型阳离子,如Be2+、Al3+等。
[相互极化作用]见离子的极化作用条。
[化学键]在分子、原子团或晶体中,相邻的两个或多个原子之间的强烈的相互作用叫做化学键。化学键有三种基本类型:离子键、共价键(包含配位键)、金属键。化学键的强度可用键能表示,一般约在125-840千焦·摩尔-1之间。
[离子键]又称电价键,系由原子得失电子后生成的阴阳离子之间通过静电作用所形成的化学键。例如氯化钠晶体中Na+和Cl-之间就是靠离子键结合在一起的。由于离子的电荷分布是球形对称的,只要空间条件许可,就可以从不同方向同时吸引几个带相反电荷的离子。离子键的特征是既没有方向性又没有饱和性。
[电价键]见离子键条。
[离子化合物]以离子键相结合而形成的化合物。如氯化钾KCl、氧化钙CaO等。离子化合物一般以离子晶体的形式存在。
[离子型分子]当离子键存在于气态分子中,这种独立的分子称做离子型分子。例如在氯化钠蒸气中,就存在着由一个Na+和一个Cl-组成的氯化钠分子NaCl。这种情况较为少见。
 [共价键]原子间通过共用电子对(电子云重叠)所形成的化学键。例如,两个氢原子共用一对电子形成氢分子H2:H:H;两个氮原子共用三对电子形成氮分子(如右图)。
[共价键]原子间通过共用电子对(电子云重叠)所形成的化学键。例如,两个氢原子共用一对电子形成氢分子H2:H:H;两个氮原子共用三对电子形成氮分子(如右图)。
共价键具有饱和性和方向性。
[共价单键]原子间共用一对电子形成共价单键。例如H2分子中的共价键。
[共价双键]原子间共用两对电子形成共价双键。例如CO2分子中,碳原子和氧原子之间以共价双键结合:
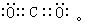
[共价叁键]原子间共用三对电子形成共价叁键。例如N2分子中的共价键。
[共价化合物]元素的原子以共价键相结合而形成的化合物。例如:水、氯化氢等。

[配位键]两原子间的共用电子对是由一个原子单独提供而形成的共价键。它和一般的共价键的区别仅在于提供电子对的方式不同。在形成配位键时电子给予体具有孤对电子,而电子接受体具有空轨道。例如氨分子NH3中N原子有一对孤对电子,而氢离子H+有空轨道,它们可以通过配位键结合形成铵离子NH4+。
[键长]表征价键性质的参数之一,是分子中两个成键原子的核间矩。一般键长越短,键就越强。
[键角]表征价键性质的参数之一,系分子中键和键之间的夹角。键角是决定分子几何构型的重要因素。例如在水分子H2O中,两个O-H键的键角是104°45′,这就决定了水分子的折线形结构。
[键能]表征价键性质的参数之一,在给定条件下,对气态物质断开1摩尔键,使成键原子变为气态原子时所需的能量。键能表示化学键的强度,键能越大,键越牢固,含有该键的分子越稳定。
[键矩]将极性键(见极性共价键)的正电荷重心作为正电中心,负电荷重心作为负电中心,所带电荷分别为+q和-q,两中心间的距离为d,则q和d的乘积就是键矩μ,用下式表示:
μ=qd
键矩是矢量,其方向定为从正电中心到负电中心,单位为德拜(Debye),用D表示。键的极性可用键矩来量度,μ愈大键的极性愈强。
[键级]分子轨道法中表示相邻的两个原子成键强度的一种数值。对双原子分子来说,把成键电子数与反键电子数的差值的一半,称为键级。在形成共价键时,成键轨道上的电子称为成键电子,它使体系的能量降低,有利于形成稳定的键;反键轨道上的电子称作反键电子,它使体系的能量升高,不利于形成稳定的键。可见,键级是衡量化学键相对强强的参数,键级愈大,键愈稳定,若键级为零,则不能成键。
[极性共价键]简称极性键。由于成键的两个原子的电负性不同,共用电子对必然偏向电负性较大的一方,使键具有极性,因此,这种键叫极性键。例如,在氯化氢HCl分子中,H-Cl键是极性键,共用电子对偏向氯原子。极性键极性的大小可用键矩来量度(见键矩条)。
[非极性共价键]简称非极性键。存在于单质分子中。由于成键的两个原子的电负性相同,共用电子对处于两核之间,没有偏移,电子云均匀分布。这种两个原子间共用电子对无偏向的共价键叫非极性共价键。例如,在H2分子、Cl2分子中的H-H键、Cl-Cl键都是非极性键。
[金属键]在金属晶体中,金属原子容易丢失电子,形成正离子,因此金属晶体中,晶格结点上存在着大量金属正离子,而在晶格中还存在着大量的“自由电子”,金属离子和自由电子之间存在的较强作用,叫金属键。金属键又称改性共价键,因为它可以看成许多原子共用许多电子的一种特殊形式的共价键,但又和共价键不同,金属键不具有饱和性和方向性。
[分子间作用力]又称范德华(Van der Waals)力。是物质分子间存在着的作用力,无方向性,无饱和性。是短程作用力,其范围约3-5×10-10米。分子间作用力比化学键的键能要弱得多,比化学键的键能小一、二个数量级。分子间作用力有取向力、诱导力和色散力三种,一般以色散力为主。分子间作用力是决定分子型物质的熔点、沸点、气化热、熔化热以及溶解度、表面张力、粘度等物理化学性质的主要因素。
[色散力]分子间作用力的主要成分。色散力普遍存在于各种分子之间。由于分子中电子不断运动和原子核的不断振动,经常发生电子云和原子核之间的瞬时相对位移,因而产生了瞬时偶极。瞬时偶极虽然存在的时间极短,但总是不断地出现。这种由瞬时偶极产生的分子间作用力叫色散力。色散力的大小和分子的变形性、电离势以及分子间的距离有关。
[取向力]分子间作用力的一种。存在于极性分子和极性分子之间。当两个极性分子相互接近时,一个分子带负电的一端要和另一个分子带正电的一端接近,使极性分子有按一定方向排列的趋势。这种由极性分子的取向而产生的分子间作用力叫取向力。取向力的本质是静电引力,取向力的大小和分子的偶极矩(见偶极矩)的大小、温度及分子间的距离有关。
[诱导力]分子间作用力的一种。存在于极性分子与非极性分子之间以及极性分子和极性分子之间;离子和分子以及离子和离子之间。当极性分子和非极性分子接近时,极性分子的偶极使非极性分子极化而产生诱导偶极,这种由于诱导偶极而产生的作用力叫诱导力。极性分子之间,离子和分子以及离子和离子之间也会产生诱导力。诱导力的大小和分子的偶极矩、变形性及分子间距离有关。
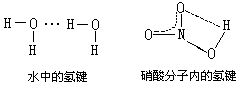
[氢键]在含氢化合物中,由一个与电负性极强的元素(如F、O、N)相结合的氢原子和另一个电负性极强带孤电子对且原子半径小的原子(F、O、N等)间产生的引力形成的。氢键的键能比化学键的键能小得多,但和范德华力相近,也可以说氢键是另一种分子间作用力,它具有饱和性和方向性。氢键可分为分子间氢键,如水中的氢键以及分子内氢键,如硝酸分子内的氢键:
[分子的极化]在外电场作用下,分子中正、负电荷中心发生分化的过程。由于外电场影响了分子中电荷的分布,使电子云与核发生了相对位移,分子发生变形,从而造成分子中正、负电荷中心的分化。非极性分子由于极化可产生极性,极性分子由于极化可使极性增强。
[偶极矩]是衡量分子极性大小的物理量。物理学中,把大小相等、符号相反、彼此相距为d的两个电荷(+q和-q)组成的体系称为偶极子,其电量与距离之积就是偶极矩(μ)。
μ=qd
偶极矩是矢量,其方向系沿两电荷连线由正到负。极性分子就是偶极子,对分子中正负电荷,可以设想它们分别集中于一点,叫正电荷中心和负电荷中心。极性分子的偶极矩等于正负电荷中心的距离乘以正电中心(或负电中心)的电量,其方向由正到负。
[永久偶极]又称固有偶极。指极性分子本身具有的偶极。这是由于极性分子中电荷分布不均匀,正电荷重心与负电荷重心不重合造成的。每个极性分子就是一个偶极子。
[固有偶极]见永久偶极条。
[诱导偶极]分子在外电场的作用下,正、负电荷重心分化而产生的偶极叫做诱导偶极。
[瞬时偶极]由于分子中电子和原子核的不断运动,经常发生电子云与核之间的瞬时位移而产生的偶极,称作瞬时偶极。这种现象是不断消失又不断发生的,时间虽短暂,但经常存在。
[八隅规则]1916年由美国物理化学家G.N.路易斯(G.N.Lewis)提出。原子相互作用形成离子或分子时,趋于形成外层为8个电子的惰性元素的构型,这通常称作八隅规则。例如,氯和钠通过得失电子形成离子化合物氯化钠,其中Na+离子和Cl-的电子排布都是惰性元素的构型。此外,在两个氯原子形成的氯气分子或一个氢原子和一个氯原子形成的氯化氢分子中原子之间通过共用电子对而形成惰性元素的电子构型。
[现代价键理论]又称电子配对理论或电子配对法,简称VB法(Valence Bond Theory)。基本要点如下:
⑴原子带有未成对且自旋相反的电子相互接近时,核间电子云密度较大,可形成稳定的化学键。
⑵共价键有饱和性,一个原子有几个未成对电子,就能和几个自旋相反的电子配对成键,未成对电子都配对成键就达到饱和。
⑶共价键有方向性,即共价键尽可能沿着原子轨道最大重叠的方向形成,这也叫做“最大重叠”原理。轨道重叠越多,电子在两核间出现的几率就越大,形成的共价键就越稳定。
现代价键理论模型只管,易于被人们接受,但在解释有些分子的形成时(如H2+、O2等)遇到了困难。这些问题由于分子轨道理论的建立才得以解决。
[电子配对法]见现代价键理论条。
[杂化轨道理论]由美国化学家鲍林(L.Pauling)及斯莱脱(J.C.Slater)在电子配对法的基础上提出。它的要点是:同一原子中能量相近的原子轨道,在成键时重新组合成一系列能量相等,成键能力更强的新轨道。这就叫原子轨道的杂化,形成的新轨道称做杂化轨道。杂化轨道理论解释了许多用电子配对法不能解释的分子的几何构型和稳定性的事实(详见有机部分)。
[分子轨道理论]又称分子轨道法(Molecular OrbitalTheory)或MO法,1932年由美国化学家密立根(R.S.Mul-liken)及德国物理学家洪特(F.Hund)提出。是现代共价键理论之一。它的要点是:从分子的整体性来讨论分子的结构,认为原子形成分子后,电子不再属于个别的原子轨道,而是属于整个分子的分子轨道,分子轨道是多中心的;分子轨道由原子轨道组合而成,形成分子轨道时遵从能量近似原则、对称性一致(匹配)原则、最大重叠原则,即通常说的“成键三原则”;在分子中电子填充分子轨道的原则也服从能量最低原理、泡利不相容原理和洪特规则。
[分子轨道]分子中描述单电子运动状态的波函数即为分子轨道,分子轨道由原子轨道线性组合而成,原子轨道形成分子轨道时遵从“成键三原则”。
[成键分子轨道]能量相近的原子轨道组合形成的分子轨道中,比原子轨道的能量有所降低的叫做成键分子轨道。在成键分子轨道中,两核间电子出现的几率增大,其能量比原子轨道中能量较低的还低,因此有利于化学键的形成。
[反键分子轨道]能量相近的原子轨道组合形成的分子轨道中,比原子轨道的能量有所升高的称反键分子轨道。在反键分子轨道中,两核间电子出现的几率减小,甚至为零,其能量比原子轨道中能量较高的还高,因此不利于化学键的形成。反键轨道通常以“*”号标记。
[成键三原则]指原子轨道组合形成分子轨道时所遵从的能量近似原则、对称性匹配原则和轨道最大重叠原则。
[单电子键]只有一个电子的共价键。如氢分子离子H+2中,两个氢原子间共用一个电子而形成的共价键。H+2中虽然没有电子配对,但却有明显的键能DH+2=269kJ·mol-1。
[三电子π键]O2分子中电子在分子轨道中的排布如下,O2[KK(σ2s)2(σ2s*)2(σ2p)2(π2p)4(π2p*)2]。除已充满的低能级的成键轨道和反键轨道基本抵消外,成键的(σ2p)2构成一个σ键;因成键的π轨道和反键的π轨道在空间方位一致,因此成键的(π2pv)2和反键的(π2p*)1构成一个“三电子π键”,成键的(π2p)2和反键的(π2p*z)1构成另一个“三电子π键”。O2的结构又可表示为:

O2分子中每个三电子π键中仍各有一个未成对电子,所以O2是顺磁性的。分子的键级:1/2(8-4)=2。
[缺电子原子]具有共价性的原子,若其价电子(见价电子条)数少于价电子层的轨道数时,称这种原子为缺电子原子。例如,硼B原子,因半径较小,电负性较大,而表现出共价性;其价电子为2s22p1,共3个,但第二电子层有4个原子轨道2s、2px、2py、2pz,硼原子就是缺电子原子。
[缺电子化合物]缺电子原子以共价键形成的不具有八隅体结构的化合物,称作缺电子化合物。例如,硼的一些化合物就是缺电子化合物,卤化硼BX3、硼烷B2H6、硼酸H3BO3等就是缺电子化合物,缺电子化合物一般可做电子接受体。
[价电子]指原子在参加化学反应时能够用于成键的电子。主族元素的价电子就是最外层电子,对过渡元素来说,价电子指最外层的s电子和次外层的d电子,甚至还有倒数第三层的f电子。
[等电子原子]在价电子层中,价轨道数等于价电子数的原子。例如,H、C、Si等原子。
[多电子原子]在价电子层中,价电子数多于价轨道数的原子。例如,N、O、S、Cl等原子。
[孤电子对]也称孤对电子。指分子或离子中某原子的最外层未共用的电子对。例如,NH3分子中,N原子有一个孤电子对;H2O分子中,O原子上有两个孤电子对:
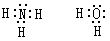
N、O原子都是多电子原子,H原子是等电子原子,因此在NH3分子和H2 O分子中都有孤电子对。
[孤对电子]见孤电子对条。
[价电子对互斥(VSEPR)理论]该理论最初由西奇威克(Sidgwiek)和鲍威尔(Powell)提出,以后吉林斯比(Gil-lespie)和宁荷姆(Nyholm)发展了这一理论。它的概念非常简单,不需要用原子轨道的概念,就能较好地判断许多共价分子的几何构型。价电子对互斥理论认为在一个共价分子中,中心原子周围电子对排布的几何构型主要决定于中心原子的价电子层中的电子对数,价电子对包括成键的σ电子对和孤电子对。分子的几何构型总是采取彼此间排斥力最小的结构,因为这样能量最低。根据价电子对互斥理论,得出理想的几何构型如下:
电子对数 几何构型
2 直线型
3 平面三角形
4 四面体
5 三角双锥
6 八面体
在分子中,孤对电子和成键电子之间静电排斥作用大小的顺序如下:
孤对-孤对>孤对-成键对>成键对-成键对
例如,在CH4 、NH3 、H2 O分子中:CH4 无孤对电子,而C-H键是相同的,所以CH4 分子是正四面体形;NH3 分子中N有一对孤对电子,它对N-H键有较大的排斥作用,所以NH3分子中N-H键之间的夹角为107°18′;H2O分子中O有两对孤对电子,它们对O-H键的排斥力更大,所以H2O分子中O-H键间的夹角是104°45′。由此可见,对简单分子AXn Er(A代表中心原子,X代表以共价键和A结合的配位体、n代表配位体的数目,E代表孤对电子,r代表孤对电子数)的几何构型就能简便地作出判断:AX4 一定是正四面体形,AX3 E一定是三角锥形,AX2 E2 一定是折线型。此外,根据具有最少数目的、角度最小的孤电子对-孤电子对排斥作用的构型最稳定的原则,还可判断其它分子的几何构型,例如,在XeF4 分子中,有六对价电子,所有价电子呈八面体形,而中心原子Xe与四个氟原子处于同一平面形成平面正方形的构型是最稳定的。
[特征电子构型]表示原子的电子层结构特性的式子,也称外围电子。根据元素原子的特征电子构型,元素周期表可划分为四个区:s区、p区、d区、f区(见周期表的分区条)。
[晶体]具有一定几何多面体形状的固体。以分子、离子或原子为质点在三维空间有规则地排列而成。晶体具有固定的熔点和各向异性。根据构成晶格质点的不同,晶体分为离子晶体、原子晶体、分子晶体和金属晶体四种类型。根据晶粒的取向不同,晶体又分为单晶体和多晶体。
[晶格]构成晶体的质点(分子、原子、离子)在空间的确定点上有规律地重复排列所构成的点阵结构。
[晶胞]晶格中含有晶体结构中具有代表性的最小部分。晶胞在空间无限地周期性的重复就成为晶格。
[晶系]根据晶胞的几何构型对晶体进行分类,晶体划分为七大晶系,一种晶系的性质用晶胞的边长(a、b、c)及夹角(α、β、γ)描述。七大晶系是:立方晶系、四方晶系、正交晶系、单斜晶系、三斜晶系、菱形晶系和六方晶系。在同一晶系中又有不同的晶格类型,共十四种晶格类型:简单立方、体心立方、面心立方、简单四方、体心四方、简单斜方、底心斜方、体心斜方、面心斜方、简单单斜、底心单斜、三斜、菱形、六方。
[无定形体]无晶体结构,内部质点在三维空间不规则排列,不具有一定几何多面体形状的固体。它和晶体不同,没有固定的熔点和各向异性。如石蜡、玻璃等。有的固体,因其结晶的取向是无序的,也称无定形态,如无定形碳,若用X-射线研究其结构可以发现它是由许多微小的石墨晶体构成的。
[离子型晶体]晶体的一种,组成晶格的质点是正、负离子。正、负离子间以离子键结合,每个离子周围被异号离子包围,它们有一定的比例关系和一定的排列顺序。例如,在氯化钠晶体中,每个钠离子被六个氯离子包围,每个氯离子被六个钠离子包围,交替延伸为整个晶体,在氯化钠晶体中不存在单个分子。离子型晶体有较高的熔点、沸点;有一定的硬度但较脆;易溶于极性溶剂,特别是水中;在熔化状态或极性溶剂中发生电离,因而具有导电性,晶体不导电。
[原子型晶体]也称共价型晶体,晶体的一种。组成晶格的质点是原子。原子间以共价键结合,结合力很强且形成空间网状结构。例如,金刚石中,碳原子形成sp3 杂化轨道,以共价键彼此相连,每个碳原子都处于正四面体的中心,组成整块晶体。在原子晶体中不存在单个小分子。原子晶体的熔点、沸点、硬度都比离子晶体高,不导电,在大多数常见的溶剂中不溶解,无延展性。常见的原子型晶体有金刚石、晶体硅、灰锡、锗、硼等单质。有一些化合物,如碳化硅SiC、氮化铝AlN、二氧化硅SiO2 等也是原子型晶体。
[金属型晶体]晶体的一种。组成晶格的质点是金属原子、金属离子和自由电子。质点间的作用力是金属键。金属晶体有三种特有的坚实结构:面心立方紧密堆积晶格、配位数是12;体心立方紧密堆积晶格,配位数是8;六方紧密堆积晶格,配位数是12。金属型晶体一般熔点、沸点高(部分低熔点金属除外,如汞Hg、镉Cd等),硬度较大,有金属光泽、良好的导热性、导电性和机械加工性能。
[分子型晶体]晶体的一种。组成晶格的质点是分子。例如,固态氧O2 、干冰CO2 ,固态氩Ar、冰H2 O等。在分子晶体内,分子间以范德华力相结合,由于结合力弱,分子晶体一般熔点、沸点低、硬度小、易挥发,通常状况下是电的不良导体。
[过渡型晶体]指介于原子晶体、金属晶体、分子晶体之间的一种过渡型。例如,石墨晶体,在晶体内碳原子以sp2 杂化形成共价键,每个碳原子与另外三个碳原子以共价键相连,六个碳原子在同一平面上形成正六边形的环,伸展形成层状结构,在同一平面的碳原子各剩一个p轨道,上有一个电子,这个电子比较自由,相当于金属中的自由电子。石墨晶体中层与层之间以范德华力结合。属于过渡型晶体的还有黑磷、云母(一种天然硅酸盐)等。
[同质多晶]同一种物质在不同条件下形成不同的结晶,称同质多晶现象。例如,单质硫在不同条件下可以形成不同的晶型。
[异质同晶]不同的物质,具有相同的晶体结构,称异质同晶现象。如氯化钾KCl、氧化镁MgO晶体都是NaCl型晶体。
[类质同晶]有一些组成不同,但化学性质类似的物质能够生成外形完全相同的晶体,称类质同晶现象,例如,明矾[KAl(SO4 )2 ·12H2 O]和铬矾[KCr(SO4 )2 ·12H2 O]都形成八面体结晶,硫酸镁(MgSO4 ·7H2 O)和硫酸镍也是类质同晶物质。
[能带]晶体中的电子,在一定能量范围内的许多能级(用彼此相隔很近的一条条水平横线表示)形成的一条带,形象地称为能带。各种晶体的能带数目和宽度都不相同。完全被电子占据的能带称作“满带”(其中的电子不能导电);部分被电子占据的能带称作“导带”(其中的电子能导电);完全未被占据的能带称作“空带”。价电子所占据的能带称作“价带”。能量比价带低的各能带一般是满带,价带可以是满带,也可以是导带。如在金属中价带是导带,故金属能导电;在绝缘体和半导体中,价带是满带,所以它们不能导电,但半导体很容易受其它因素的影响,使价带中的电子数目减少,或使空带中出现一些电子而成为导带,因而也能导电。
[等电子体]具有相同电子数的分子或离子称等电子体。例如,都含有14个电子的CO和N2 两种分子,就是等电子体。在一般情况下,等电子体在分子轨道中的电子排布和成键情况是相似的,这一规律称作等电子原理。由于等电子体具有相似的结构,所以在性质上也有许多相似之处。如:
CO N2
分子量 28 28
熔点(K) 73 63
沸点(K) 83 77
液体密度(g·cm-3) 0.793 0.796
另外,如NO3- 和CO32- 也是等电子体或称等电子离子,它们的几何构型都是平面三角形;PO43- 、SO42-、ClO4- 三种等电子离子的几何构型都是正四面体。
[等电子原理]见等电子体条。
[等离子体]亦称等离子态。气体被加热到很高的温度或被辐射后,使原子呈电离态,整个气体将成为带正电的离子和带负电的电子所组成的集合体,而且正负电量相等,这就叫做等离子体。象火焰和电弧中的高温部分,太阳和其它恒星的表面气层等都是等离子体。
[元素周期律]指元素性质随原子序数的递增呈周期性变化的规律。元素性质的周期性变化实质上是由原子的电子层结构的周期性变化决定的。元素周期律是1869年俄国化学家门捷列夫(Meндeлeeв)在前人研究成果的基础上提出的,当时认为,元素的性质随着原子量的递增而呈周期性变化。现在,根据现代原子结构理论对元素周期律的本质有了更深刻的认识,这样才把元素周期律修正为现在的形式,同时对元素周期表也作了许多改进。
[元素周期系]根据元素周期律把化学元素组织在一起所形成的完整体系,叫做化学元素周期系。
[元素周期表]根据元素周期律,把现在已知的107种元素中电子层数目相同的各种元素,按原子序数递增的顺序从左到右排成横行,再把不同横行中最外电子层的电子数相同的元素(或外围电子构型相似的元素)按电子层数递增的顺序由上而下排成纵行。这样得到的表,叫元素周期表。元素周期表是元素周期律的具体表现形式,它反映了元素之间相互联系的规律。
[原子序数]元素周期表中按元素核电荷数由小到大的顺序给元素编号,这种序号叫做元素的原子序数。原子序数在数值上等于元素的核电荷数(即质子数)。
[周期]在元素周期表中,具有相同电子层数又按照原子序数递增的顺序排列的每一横行中的一系列元素。元素周期表中共分7个周期,第1周期,含两种元素;第2、3周期各含八种元素;第4、5周期各含18种元素;第6周期有32种元素;其中,含元素较少的第1、2、3周期叫短周期;含元素较多的第4、5、6周期叫长周期;第7周期到现在只发现了21种元素,还没有填满,叫不完全周期。
[短周期]见周期条。
[长周期]见周期条。
[不完全周期]见周期条。
[族]元素周期表中的纵行。周期表中有18个纵行。除第8、9、10三个纵行叫做第Ⅷ族外,其余15个纵行,每个纵行为一族。周期表中有七个主族、七个副族,一个零族和第Ⅷ族。族序号习惯用罗马数字表示。由短周期元素和长周期元素共同组成的族叫主族,族序号后面标一A字;只由长周期元素构成的族叫副族,族序号后面标一B字;惰性元素叫做0族。
[主族]见族条。
[副族]见族条。
[零族]见族条。
[第Ⅷ族]周期表中第8、9、10三个纵行的元素为第Ⅷ族,其中包括铁Fe、钴Co、镍Ni、钌Ru、铑Rh、钯Pd、锇Os、铱Ir、铂Pt,共九种元素。
[铁系元素]第Ⅷ族元素中铁、钻、镍这三种元素的性质很相似,称为铁系元素。
[铂系元素]第Ⅷ族中钌、铑、钯、锇、铱、铂六种元素性质相似,称为铂系元素。
[镧系元素]周期表中第六周期ⅢB族中57-71号的15种元素称作镧系元素。其中包括镧La、铈Ce、镨Pr、钕Nd、钷Pm、钐Sm、铕Eu、钆Gd、铽Tb、镝Dy、钬Ho、铒Er、铥Tm、镱Yb、镥Lu,共十五种元素。
[锕系元素]周期表中第七周期ⅢB族中89-103号的15种元素称作锕系元素。其中包括锕Ac、钍Th、镤Pa、铀U、镎Np、钚Pu、镅Am、锔Cm、锫Bk、锎Cf、锿Es、镄Fm、钔Md、锘No、铹Lr,共十五种元素。它们都是放射性元素。其中只有锕、钍、镤、铀四种元素存在于自然界,在铀以后的元素称为超铀元素,它们都是用人工核反应合成的,极微量的镎Np和钚Pu也存在于自然界中。
[超铀元素]见锕系元素条。
[稀土元素]周期表中第ⅢB族中钪Sc、钇Y和镧系元素在性质上都非常相似,并在矿物中共生。18世纪时,人们习惯上把不溶于水的固体氧化物叫做“土”。19世纪初,稀土元素被发现时,它们很象“土”。又因是在很稀有的矿物中发现的,因此把它们叫“稀土”。稀土元素并不稀有,在自然界中约有250多种稀土矿。稀土元素主要应用于玻璃制造、陶瓷工业、冶金工业、原子能工业、电子技术以及用作催化剂等。
[过渡元素]广义的过渡元素包括周期表中所有副族元素和第Ⅷ族元素。也有的将ⅠB、ⅡB两个副族除外。过渡元素都是金属,在现代科学技术中有重要应用。
[内过渡元素]镧系和锕系元素的总称。
[元素周期表的分区]根据原子的电子层结构特征,元素周期表可划分为四个区。S区:包括ⅠA和ⅡA两个主族,特征电子构型分别为ns1 和ns2 ;p区:包括ⅢA-ⅦA五个主族和0族,除He以外,特征电子构型为ns2 np1-6;d区:包括ⅠB-ⅦB七个副族和第Ⅷ族,最外层有两个电子,次外层有1-10个d电子,特征电子构型为(n-1)d1-10 ns2;f区:包括镧系和锕系,最外层有2个s电子;次外层电子排布基本相同,有2个s电子和6个p电子(个别有d电子),倒数第三层有 1-14个f电子,特征电子构型一般是(n-2)f1-14 ns2 。其中,d区元素和f区元素都是过渡元素。
[同位素]原子核内质子数相同而中子数不同的同一种元素的不同原子互称同位素。同位素中有的是稳定的,称稳定同位素,在天然元素中多数都有稳定同位素,也有一些元素尚未发现稳定同位素,如氦、氟、钠、铝、磷、金等;有的同位素具有放射性,称放射性同位素,放射性同位素中除天然放射性同位素外,还有人工放射性同位素。天然存在的同一元素的各种同位素均匀地分布在自然界各种物质中。同一元素的各种同位素的原子核虽有差别,但核外电子排布相同,化学性质基本相同。
[核素]具有一定质子和中子数目的一种原子称为核素。例如,氧有三种核素,它们核内的质子数都是8,而中子数分别是8、9、10,质量数分别是16、17、18,氧的这三种核素就是氧的同位素:168O、178 O、188O。
[同量素]又称同量异序素。质量数相同而核内质子数不同的各种不同元素的原子互为同量素。例如,4018Ar、4019 K、4020Ca互为同量素。
[道尔顿原子学说]1803年由英国化学家道尔顿(JohnDalton,1766-1844)提出,其要点如下:一切物质都是由极小的微粒--原子所组成,原子不能自生自灭,也不可再分;同种物质含有同种原子,不同物质含有不同种原子,不同种原子具有不同的大小、性质和质量(即原子量);单质由简单原子组成,化合物则由复杂原子所组成(为数不多的简单原子可组成复杂原子)。1808年道尔顿在其著作《化学哲学新体系》中全面阐明了这一学说,在化学发展史上是一个重要的里程牌。这一学说的不完善之处在于,没有提出分子概念,未把原子与分子加以区别,并认为原子是不可再分的微粒。这些问题在分子学说提出后以及进入20世纪以后关于原子结构的一系列新发现和原子结构理论建立后才得到解决。
[原子分子学说]又称原子分子论,是在道尔顿原子论和阿佛加德罗分子论的基础上,经意大利化学家康尼查罗(Stanislao Cannizzaro,1826-1910)的实验论证,于1860年建立的。其要点是:一切物质都是由分子组成的,分子是由原子组成的;分子是保持物质化学性质的最小单位,原子则是化学反应中不可再分的最小微粒;单质分子由同种原子组成,化合物分子由不同原子组成,分子质量等于组成它的原子质量的总和。原子分子学说的建立,是化学发展史上的重要阶段。
[X-射线]又称伦琴射线,1895年由德国物理学家伦琴(Roentgen)发现。这是一种波长很短的电磁波,波长约为20-0.06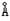 (1=10-10米),大致介于紫外线和γ射线之间。X-射线有一定的穿透能力,还能使荧光物质发光、照相底片感光、气体电离等。X-射线应用很广,在科学研究中用于研究晶体结构,工业上用于金属探伤,医疗上用于人体透视。
(1=10-10米),大致介于紫外线和γ射线之间。X-射线有一定的穿透能力,还能使荧光物质发光、照相底片感光、气体电离等。X-射线应用很广,在科学研究中用于研究晶体结构,工业上用于金属探伤,医疗上用于人体透视。
[α-射线]放射性原子核所发出的α粒子流(α粒子即氦原子核)。α-射线中,α粒子的动能很大,可达几兆电子伏特以上;但由于α粒子的质量比电子大得多,通过物质时极易使其中的原子电离而损失能量,所以它的穿透物质的本领比β射线弱得多,容易被薄层物质阻挡。
[β-射线]放射性原子核所发出的电子流(或正电子流)。电子的动能可达几兆电子伏特以上,由于电子的质量小,速度大,通过物质时不易使其中的原子电离,所以它的能量损失较慢,穿透物质的本领比α射线强得多。
[γ-射线]波长极短[通常指1(10-10米)以下]的电磁辐射。原子核在衰变过程中能放出γ-射线。
[d-pπ键]是一种特殊形式的π键。指某元素的原子用d轨道与另一原子具有相同对称性的p轨道形成的π键。如第三周期的一些元素在和O、F、Cl等原子结合时就可能形成这种形式的键。现代研究表明,在SiF4、PF3、PO43-、SO42-、ClO4- 等分子和原子团中都存在d-pπ键,其中做为中心原子的 Si、P、S、Cl等原子用空的3d轨道和 F、O等原子的充满电子的p轨道成键。
[非极性分子]指没有偶极的分子,即分子的固有偶极为零的分子。这种分子里负电荷中心与正电荷中心是相重合的。如H2分子、Cl2分子、CO2分子、CH4分子、CCl4分子等。
[极性分子]指具有固有偶极的分子。这种分子里负电荷中心与正电核中心不重合,而显示出极性,如HCl分子、H2O分子、NH3分子、CHCl3分子等。分子极性的大小用偶极矩来度量。
[物理变化]物质的外形或状态发生改变而没有新物质生成的变化。物质发生物理变化时,仅改变了其物理性质(如聚集状态、密度、溶解度、电导率等),而不改变其化学组成和化学性质。例如水的蒸发和凝固、蔗糖溶解等。物质发生物理变化后,可以通过物理方法使它恢复到原来状态,例如液态水降低到0℃以下时变为冰,再将温度升高到0℃以上时冰又熔化成水。
[化学变化]又称化学反应。是指在原子核组成不变的情况下生成新物质的变化。这是由于核外电子运动状态的改变而引起的物质组成的质变。例如碳的燃烧、铁生锈、石灰石受热分解等。化学变化以质变为其特征,同时还伴随有能量的变化,表现为反应过程常有放热、吸热等现象。有时反应结果出现变色、放出气体、生成沉淀等,这些现象有助于人们判断化学变化的发生。化学变化一般分为化合反应、分解反应、置换反应和复分解反应等。
[化学反应]见化学变化条。
[物理性质]物质本身的一种属性,不需要发生化学变化就表现出来的性质。例如状态、颜色、气味、密度、硬度、熔点、沸点、蒸气压、导热性、导电率、延性、展性等。这些性质都没改变物质的化学组成。
[化学性质]物质本身所具有的、只有在化学变化中才能表现出来的性质。例如酸性、碱性、氧化性、还原性、化学稳定性等。物质的化学性质主要决定于物质的组成和结构。例如,Na、Mg、Al的还原性依次减弱,是由于它们的最外层电子数目依次增多,原子半径依次减小,核电荷数依次增多,造成了它们在化学反应中失电子能力依次减弱。Na、Mg、Al的还原性只有在化学反应中才能表现出来。
[分解反应]化学反应的基本类型之一。是一种物质生成两种或两种以上物质的反应。例如:
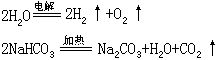
在分解反应中,如果反应前后各元素的化合价保持不变,就属于非氧化-还原反应,如NaHCO3受热分解;如果有些元素的化合价发生改变,就属于氧化-还原反应,如电解水的反应。
[化合反应]化学反应的基本类型之一,是两种或两种以上的物质生成另一种物质的反应。例如:
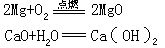
在化合反应中,如果反应前后各元素的化合价保持不变,就属于非氧化-还原反应,如氧化钙与水化合;如果元素的化合价发生改变,就属于氧化-还原反应,如镁条在空气中燃烧。在化合反应中,反应物越活泼,生成物就越稳定,反应就越容易进行。
[置换反应]化学反应的基本类型之一,是一种单质跟一种化合物反应生成另一种单质和另一种化合物的反应。例如:
2Na+2H2O=2NaOH+H2↑
Fe+CuSO4=FeSO4+Cu
Cl2+2KI=2KCl+I2
在置换反应中,反应前后某些元素的化合价必定改变,因此置换反应都属于氧化-还原反应。置换反应中,按化学活动性顺序,较活动的金属能跟酸或盐反应,置换出较不活动的金属和氢气;较活动的非金属能与盐反应,置换出较不活动的非金属。
[复分解反应]化学反应的基本类型之一。是两种化合物互相交换成分生成另外两种化合物的反应。例如:
HCl+NaOH=NaCl+H2O
FeS+2HCl=FeCl2+H2S↑
AgNO3+NaCl=AgCl↓+NaNO3
在复分解反应中,反应前后元素化合价必定不变,因此复分解反应都属于非氧化-还原反应。从复分解反应的产物中有无气体、沉淀、弱电解质(如水)的生成,来判断反应是否能发生,只要生成物有气体、沉淀、弱电解质三者之一的,复分解反应就可以趋于完成。
[中和反应]酸和碱相互作用生成盐和水的反应。例如盐酸和氢氧化钠反应。
HCl+NaOH=NaCl+H2O
强酸和强碱的反应,实质上是酸中的氢离子与碱中氢氧根离子相互结合生成难电离的水分子的反应。
H++OH-=H2O
[水合反应]又称水化反应。物质跟水发生的化合反应。一般指分子或离子与水结合成水合物或水合离子的过程。例如,
CuSO4+5H2O=CuSO4·5H2O
H++H2O=H3O+
在有机化学中也指有机物分子中不饱和键(双键或三键)在催化剂作用下和水分子化合的反应。
[水化反应]见水合反应条。
[离子反应]电解质在溶液里所发生的离子间的反应。有两类情况:
⑴电解质在溶液里发生的离子互换反应,属于非氧化-还原反应,反应发生的条件是生成物之一是难溶物质,易挥发性物质或极难电离的物质,这类离子反应的特征一般是朝着减小离子浓度的方向进行。例如:
Ag++Cl-=AgCl↓
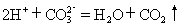
⑵属于氧化-还原反应的离子反应,例如:
Zn+2H+=Zn2++H2↑
Cl2+2I-=2Cl-+I2

这类离子反应的发生条件遵循氧化-还原反应规律。
[氧化反应]简称氧化。物质(分子、原子或离子)失去电子或电子偏离的反应。在氧化反应中物质所含的元素化合价(或氧化数)升高,这是判断氧化反应的依据。氧化反应和还原反应相伴而生。例如:

[还原反应]简称还原。物质(分子、原子或离子)得到电子或电子对偏近的反应。在还原反应中物质所含的元素化合价(或氧化数)降低,这是判断还原反应的依据。还原反应和氧化反应相伴而生。参见氧化反应。
[氧化-还原反应]物质发生电子转移(或电子对偏移)的反应。在氧化-还原反应里,一种物质失去电子(或电子对偏离),必然同时有另外的物质得到电子(或电子对偏近),而且得失电子的总数相等。在氧化-还原反应里氧化和还原总是同时发生的。反应前后元素的化合价(或氧化数)发生变化是氧化-还原反应的特征。例如:
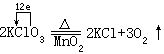
[氧化剂]在氧化-还原反应中得到电子(或电子对偏近)的反应物。在反应中氧化剂中元素的化合价(或氧化数)降低。氧化剂能氧化其它物质而自身被还原,例如:
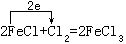
Cl2是氧化剂,在反应中获得电子,化合价(或氧化数)降低,将FeCl2氧化成FeCl3而Cl2自身被还原成Cl-。氧化剂通常是指容易获得电子的物质,常见的有:
⑴活泼的非金属单质、如氧、氯、溴等。
⑵具有高化合价(氧化数)的金属离子,如Fe3+、Sn4+等。
⑶含有高化合价(氧化数)元素的含氧化合物,如KMnO4、K2Cr2O7、MnO2、KClO3、HNO3、H2SO4(浓)、PbO2等。
⑷过氧化物,如H2O2、Na2O2、BaO2等。
根据氧化剂获得电子的难易程度,可分为弱氧化剂和强氧化剂,定量地判断氧化剂氧化能力的大小,应依据氧化剂及其还原产物所构成的氧化还原电对的标准电极电位E0值来确定,E0值越正,表明氧化剂的氧化能力就越强。
[还原剂]在氧化-还原反应中失去电子(或电子对偏离)的反应物。在反应中还原剂中元素的化合价(或氧化数)升高。还原剂能还原其它物质而自身在反应中被氧化。还原剂通常是指容易失去电子的物质,常见的有:
⑴活泼金属,如钠、钾、镁、铝、铁等。
⑵具有低化合价(氧化数)的金属离子,如Fe2+、Sn2+等。
⑶非金属离子,如I-、S2-等。
⑷含有低化合价(氧化数)元素的含氧化合物,如CO、SO2、Na2SO3、Na2S2O3、NaNO2等。
根据还原剂失去电子的难易程度,可分为弱还原剂和强还原剂,定量地判断还原剂还原能力的大小,应根据该还原剂及其氧化产物所组成的氧化还原电对的标准电极电位E0值来确定,E0值越负,表明该还原剂的还原能力越强。
[氧化性酸]一般指中心原子获得电子显示氧化性的酸。如浓硫酸、硝酸、氯酸、高氯酸、高锰酸、重铬酸等,其中心原子在氧化-还原反应中容易获得电子而显示氧化性,可用作氧化剂。任何酸的水溶液中都不同程度地电离出H+,H+在一定条件下可获得电子而形成H2,这也是酸的氧化性的表现,实质上是H+的一种性质。应该与酸的中心原子获得电子所呈现的氧化性区别开来。通常把盐酸、稀硫酸等称作为非氧化性酸。
[歧化反应]也称自身氧化-还原反应。是氧化-还原反应的一种类型。在歧化反应中,同一种元素的一部分原子(或离子)被氧化,另一部分原子(或离子)被还原。这是由于该元素具有高低不同的氧化态,在适宜的条件下向较高和较低的氧化态转化的缘故。例如:
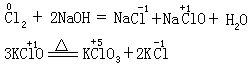
[自身氧化-还原反应]见歧化反应条。
[非氧化-还原反应]反应过程中,反应物的原子或离子不发生氧化数变化的反应,例如复分解反应和络合反应等(参见氧化-还原反应条)。
[燃烧]伴有发光放热现象的剧烈氧化反应。燃烧可分为完全燃烧和不完全燃烧两种,燃料不完全燃烧的产物往往会造成对大气的污染,还会降低热能的利用率,因此要设法使燃料燃烧完全。一般将固体燃料粉碎,液体燃料喷成雾状来增加和空气的接触机会,同时采取通风等措施,以提高燃料的利用率。
[着火点]又称燃点。物质开始着火时的温度。物质燃烧前,必须加热到着火点,物质开始燃烧后,释放出热量,就不需要再从外界获得热量。固体燃料的着火点不是固定不变的,一般颗粒越细,表面积越大,着火点就会降低,如炭粉与炭块的着火点是不同的;液体燃料的着火点与燃料雾化程度有关。纯气体燃料在常压下的着火点是一定的,如一氧化碳的着火点为650℃、氢气的着火点为585℃、甲烷的着火点为537℃等。
[燃点]见着火点条。
[闪点]又称内燃点。表示可燃性液体性质的指标之一。是液体表面的蒸气和空气的混和物与火接触初次发生蓝色火焰闪光时的温度。用标准仪器测定闪点,仪器有开杯式和闭杯式两种,一般前者用于测定高闪点液体的闪点,后者用于测定低闪点液体的闪点。闪点比着火点的温度低些。可燃性液体的闪点和着火点表示其发生爆炸或火灾的可能性大小,这对可燃性液体的运输、储存和使用的安全等有极大的关系。
[闪燃点]见闪点条。
[缓慢氧化]是氧化反应的一种形式。反应过程比较缓慢,不象燃烧那样剧烈地放热发光。例如铁生锈、呼吸作用、食物的腐败等,都是缓慢氧化的实例。
[自燃]由缓慢氧化而引起的自发燃烧。可燃物在缓慢氧化过程里也产生热量,在某种情况下热量不易散失,以致越积越多,温度逐渐升高,达到那种可燃物的着火点时不经点火就会自发地燃烧起来。例如稻草、麦杆、煤屑、沾有油的布等大量堆积在不通风的地方都有可能自燃。
[爆炸]物质发生的变化不断急剧增速并在极短时间内放出大量能量的现象。爆炸主要由化学反应或核反应所引起。爆炸发生时,温度和压力突然升高,产生爆破作用或推动作用。爆炸广泛应用于开矿、筑路、推动发动机等。某些可燃物在有限空间里发生急速燃烧的时候,常会发生爆炸。所以油库附近和面粉工厂都严禁烟火。
[爆炸极限]指一种可燃气体、蒸气或粉末和空气的混和物能发生爆炸的浓度范围。例如空气中混有氢气、汽油蒸气、面粉等时,在一定浓度范围内,遇到火花就会使火焰蔓延而发生爆炸。其最低浓度称为下限(或低限),最高浓度称为上限(或高限),浓度低于或高于这一范围,都不会发生爆炸。爆炸极限通常用可燃性物质在爆炸混和物中体积百分比表示,例如氢气的爆炸极限为4.1-74.2%,4.1%为下限,74.2%为上限。有时也用每立方米混和物中所含可燃性物质的质量(克)来表示,这种表示方法,大多用于可燃性粉尘的爆炸极限,例如铝粉的爆炸极限为40克/米3,这是铝粉的爆炸下限,一般不计可燃性粉尘的爆炸上限。
[火焰]可燃性气体燃烧时所发生的现象。是燃烧的一种特殊情况,火焰是指气态可燃物在燃烧时形成的一个发光、放热的区域。液态或固态可燃物在燃烧时,如能先变成气体,燃烧时也会有火焰生成。
火焰可分为三个部分:
⑴内层,带蓝色,因供氧不足,燃烧不完全,温度低,有还原作用,称为内焰或还原焰;
⑵中层,明亮,温度较内层高;
⑶外层,无色,因供氧充足,燃烧完全,温度最高,有氧化作用,称为外焰或氧化焰。
对有灯芯的火焰,如蜡烛和酒精灯火焰则可分为:
⑴焰心,中心黑暗部分,由能燃烧但还未燃烧的气体(蒸气)所组成;
⑵内焰,包围焰心的最明亮部分,是气体未完全燃烧的部分,含有碳粒子(或其它固体粒子),被灼热发出强光,并有还原作用,也称还原焰;
⑶外焰,最外面几乎无光的部分,是气体完全燃烧的部分,含有过量而强热的空气,有氧化作用,也称氧化焰。
[氧化焰]见火焰条。
[还原焰]见火焰条。
[吸附]固体或液体表面对气体或溶质的吸着现象。由于分子间作用力相互作用而产生的吸附为物理吸附,如活性炭对气体的吸附。物理吸附一般在常温下发生,吸附速度快,吸附热小,吸附无选择性。由于化学键的作用而产生的吸附为化学吸附,如镍催化剂吸附氢气,化学吸附有化学键的破坏与生成,放出的吸附热较大,所需的活化能也较大,化学吸附需在高温下进行,并有选择性。
[物理吸附]见吸附条。
[化学吸附]见吸附条。
[吸附质]被吸附剂所吸附的物质。例如,在制糖工业中,常利用活性炭的吸附作用来脱色,被吸附的有色物质即为吸附质。
[吸附剂]通常指对气体或溶液中的微粒具有吸附作用的固体物质。一般要求具有较大的吸附表面和选择性吸附的能力。如活性炭、硅胶、活性氧化铝、硅藻土等都是常用的吸附剂。明矾水解生成的氢氧化铝胶体,也可看作是吸附剂。
[解吸]是与气体的吸附或吸收相反的过程。如用活性炭吸附二氧化氮后,当加热或降低压力时可使二氧化氮逸出。
[风化]在室温和比较干燥的空气里,结晶水合物失去一部分或全部结晶水,使原有结晶形状转变或破坏的一种现象。例如,在室温时,碳酸钠晶体(Na2CO3·10H2O)放在干燥空气里,会逐渐失去结晶水变成粉末。
[潮解]某些晶体自发地吸收空气里的水蒸气,在其表而逐渐形成饱和溶液的现象。例如无水氯化钙、氯化镁和固体氢氧化钠在空气中潮解。潮解是由于该化合物饱和溶液的蒸气压低于同温下空气中的水蒸气的分压。因而不断吸收水分而潮解。
[结晶水]许多物质从水溶液中形成晶体析出时,晶体里常结合一定数目的水分子。这样的水分子叫做结晶水。如胆矾(CuSO4·5H2O)中就含有结晶水。
[结晶水合物]含有结晶水的物质叫做结晶水合物。又称水合物、水化物。如绿矾(FeSO4·7H2O)、二水合硫酸(H2SO4·2H2O)、六水合氯化铁(FeCl3·6H2O)等。除水合离子外,结晶水合物大多是晶态物质。结晶水合物在受热时会失去结晶水,并有显著的吸热效应。
[重结晶]又称再结晶。将晶体溶解或熔化,然后又重新从溶液或熔体中结晶出来的过程。重结晶可减少或除去晶体中的可溶性杂质,是提纯物质的一种重要方法。重结晶还可以细化晶粒,是改变某些金属和合金的晶体结构以改善其性能的一种方法。
[再结晶]见重结晶条。
[沉淀]由于化学反应而生成溶解度较小的物质从溶液中析出的过程。也指沉淀过程中析出的固体物质。在化学实验和生产实践中广泛地应用沉淀方法进行物质的分离。例如沉淀法是定量分析中的一种重要的分离方法。
[陈化]沉淀析出以后,让初生的沉淀与原溶液在一起继续放置一段时间的过程。经过陈化作用,可以使微小的晶体溶解,而较大的晶体长得更大,使沉淀颗粒变大,易于过滤,而且变得较为纯净,但是,当溶液中有混晶共沉或伴随有后沉淀时,则不一定能提高沉淀的纯度。
[过滤]一种分离含有不溶性固体的液体或气体混和物的操作。一般用普通玻璃漏斗、布氏漏斗、热过滤漏斗做为过滤器,用滤纸、滤布、砂层做为过滤介质,将混和物进行过滤,得到的澄清液体称滤液,留在过滤介质上面的固体颗粒叫滤渣。有常压过滤、减压过滤和热过滤等方法。过滤在科研与生产中有广泛的应用。
[蒸馏]利用液体混和物中各组分的沸点不同,以分离各个组分、纯化物质的一种操作。将混和物加热至沸腾,使其中某种组分气化,然后将其蒸气冷凝为液体。蒸馏可使原混和物中各组分得到部分或完全分离。蒸馏的方法很多,有简单蒸馏、真空蒸馏、精馏、恒沸蒸馏等。主要用于物质的精制和混和物的分离。例如清除天然水中的杂质制取蒸馏水等。
[蒸发]在液体表面发生的气化现象。在任何温度下都能进行蒸发。蒸发时,液体必须从其周围吸收热量,所以温度愈高,蒸发愈快。各种液体的挥发度不同,因而在相同条件下,各种液体的蒸发速度不同,例如乙醇的蒸发速度比水快,海水中水的蒸发速度比淡水慢。在生产中,蒸发是指在对溶液加热过程中,一部分溶剂气化,使溶液浓缩或使溶质析出晶体的过程。蒸发的必需条件是不断地供给热能和不断地排除气化了的蒸气。为了加快蒸发的速度,可采用沸腾蒸发、减压蒸发等。
[分馏]又称精馏,是蒸馏方法的一种。在一个设备内同时进行多次部分气化和部分冷凝,以分离液体混和物的组分。适合于沸点相差很小的互溶的两种或两种以上的液体组分所组成的混和物的分离,例如石油的分馏。操作时,使液体混和物在蒸馏釜中加热蒸发,蒸气进入分馏塔,部分冷凝回流,使与从蒸馏釜连续上升的蒸气密切接触,可得到与重复简单蒸馏若干次相当的效果,从而提高了各组分的分离程度。分馏可同时得到几种馏出液。
[干馏]固体燃料的热化学加工方法之一。如煤、木材、油页岩等在隔绝空气下加热,使之分解生成固态的焦炭、液态的焦油和气态的煤气等产物。根据加热的最终温度,一般可分为高温干馏(约900-1100℃)、中温干馏(约660-750℃)和低温干馏(约500-580℃)。
[稀释]加溶剂于溶液中以降低溶液浓度的过程。稀释时常有放热或吸热现象发生,称为稀释热。
[浓缩]减少溶液中溶剂的量以提高溶液浓度的过程。浓缩是常用的脱水作业之一。有时,在浓缩过程中,液体中的溶质析出发生沉淀,以利于进一步的脱水(过滤或干燥)。
[脱水]从物质中除去水分的过程。一般有三种不同的方式:
⑴除去或降低物料中的不定量的水分,例如食物的脱水。
⑵除去结晶水合物中的结晶水,例如胆矾加热得到无水硫酸铜和水。
⑶在脱水剂或催化剂存在下,使化合物分子中(或分子间)的羟基和氢原子以水的形式脱去,例如乙醇在浓硫酸作用下,加热到140℃生成乙醚和水,加热到160℃以上生成乙烯和水。
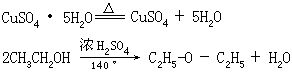
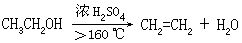
[脱水性]能使有机化合物分子中的羟基与氢原子以水分子形式脱去的性质。例如浓硫酸与甲酸混和加热时,甲酸脱水生成一氧化碳和水,因此浓硫酸有脱水性。
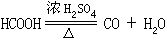
[吸水性]能除去或减少物质中所含不定量的湿存水的性能。例如浓硫酸、碱石灰能吸收空气中的水分,因此浓硫酸具有吸水性。
[干燥]指除去附在固体、气体或混在液体内的游离水(或溶剂)的过程。可采用干燥剂、加热、减压等方式来干燥。
[干燥剂]能吸收潮湿物质中不定量的湿存水的物质。通常有两类:
⑴化学干燥剂,吸收水分时伴随有化学反应发生,如无水氯化钙、浓硫酸、碱石灰等。
⑵物理干燥剂,吸收水分而无化学反应发生,如硅胶吸收空气中的水分。
不同干燥剂的干燥能力不同,测定办法是:先把被水蒸气饱和的空气在25℃时以1-3升/小时的速度通过定量的干燥剂,然后再测定被干燥的空气中剩余水分的含量,剩余的水分越少,说明干燥剂的吸水性越好。
[萃取]利用不同的物质在选定的溶剂中溶解度的不同,分离混和物中的组分的方法。习惯上指用溶剂分离液体混和物中的组分,即液-液萃取(又叫溶剂萃取);也可用溶剂分离固体混和物中的组分,即固-液萃取(又叫浸取)。液-液萃取是定量分析中分离方法的一种,主要用于元素的分离和富集。
[萃取剂]萃取所用的溶剂。必须具备对液体或固体混和物中的组分具有选择性溶解的能力。同时应具有较好的热稳定性和化学稳定性,以及较小的腐蚀性和毒性。例如,碘的水溶液用四氯化碳萃取,极大部分的碘进入四氯化碳层而与其它物质分离。四氯化碳为萃取剂。
[分配定律]溶质同时分别溶解在两种互不相溶或几乎互不相溶的混和液体中,它在两种液体中的浓度比等于它在两种液体中的溶解度之比。利用这个规律可以用一种溶剂把另一种溶液中的溶质萃取出来。如用跟水不相溶的四氯化碳作溶剂,可以从碘水中把碘萃取出来。
[凝华]气态物质不经过液态而直接凝结为固态的过程。如水蒸气能直接凝结于物体表面成为霜,硫蒸气遇冷直接生成硫华。凝华时有热量放出。
[升华]固体不经过液态直接转变成为气态的过程。在三相点以下的任何温度,当外界压力低于该物质平衡蒸气压时,固体就会升华。例如,在气温低于0℃时(水的三相点为0.01℃),空气中水蒸气的分压低于其平衡蒸气压,冰或雪就会因为升华而消失。又如,碘的三相点为115℃,三相点的蒸气压为12kPa,故碘很容易升华,将碘晶体放在试管中微热,即可看到紫色碘蒸气生成。但如加热温度高于115℃(碘的三相点)时,固体碘就会熔化。
[钝化]某些金属经化学方法(如跟强氧化剂反应)或电化学方法(如阳极氧化)处理,由活泼态变为不活泼态(即钝态)的过程。由于钝化后,金属表面形成致密的氧化物保护薄膜,因而不易腐蚀。例如,铁能溶于稀硝酸,但铁在浓硝酸中浸过后,因铁钝化不再溶于稀硝酸。
[化学腐蚀]由于金属跟它周围的物质直接起化学反应,从而造成金属被腐蚀的过程。在化学腐蚀中,跟金属起化学反应的多是氧气、硫化氢、二氧化硫等气体,生成相应的氧化物和硫化物等。有些金属,由于化学腐蚀的结果常在表面上形成一层致密的薄膜,从而保护了金属不再继续被腐蚀。如铝在空气中很快被氧化,表面形成一层致密Al2O3薄膜,起到了保护金属的作用。
[气焊]使乙炔(或其它可燃气体)在氧气不足的情况下燃烧:
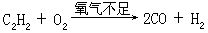
产生具有还原性的一氧化碳和氢气,它们继续与氧化合产生二氧化碳与水,伴随着产生高温,使金属焊条熔化填满缝隙而使金属焊接起来。火焰的还原性对焊接金属极为有利,防止在焊接时金属被氧化,使焊缝被氧化物沾污而造成焊接不牢的现象发生。
[气割]利用氧炔焰加热时使用过量的氧气,让熔化的金属进而被氧化,在工作物上形成一条割缝,从而将金属割断。过量的氧气是从附加的另外一个氧气吹管里吹出的。
[固氮]使空气中游离的氮气转变为含氮化合物的过程。如工业上在高温、高压和催化剂作用下,将氮气和氢气合成氨;土壤中的固氮菌在常况下,靠固氮酶的作用,将空气中的氮还原为氨;自然界的雷电作用使氮气转化为一氧化氮,并进一步氧化为二氧化氮等。科学上关于模拟生物固氮的研究,将为人工固氮开辟新途径。
[漂白]除去纤维纺织材料和纸浆等中所含有色物质的过程。通常是氧化作用,有时是还原等其它作用。漂白棉、麻、人造纤维和纸浆时,一般用漂白粉、漂白精、次氯酸钠、亚氯酸钠和过氧化氢等。漂白蚕丝和羊毛,可用过氧化氢,有时也用二氧化硫,但漂得的白色物质不够稳定,容易发黄。对油脂的漂白通常采用漂白土,其实质是吸附脱色作用,也称为漂白。
[光化反应]见光化作用条。
[光化作用]又称光化反应。物质由于光(一般为可见光或紫外线)的作用而引起的化学反应。例如,碳水化合物的合成,染料在空气中褪色,胶体的感光作用等,范围很广泛,可能是化合、分解、氧化-还原等化学反应,主要分光合作用和光解作用两类。
[光合作用]光化作用的一种。一般指二氧化碳和水,在日光照射下,借植物叶绿素的帮助,吸收光能合成碳水化合物的过程。可以表示如下:
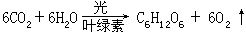
有时也包括在光的作用下,合成蛋白质、脂肪等有机物的过程。
[光解作用]光化作用的一种。物质由于光的作用而分解的过程。例如碘化氢在紫外线的照射下,吸收光能分解成碘和氢气,卤化银、浓硝酸见光分解等。
[蜕变]原子核自发地放射出一个α或β粒子、同时自身转变为另一种原子核的过程。常称为α衰变或β衰变。
[核反应]某种微观粒子与原子核相互作用时,使核的结构发生变化形成新原子核,并放出一个或几个粒子的过程。重核可以发生裂变。历史上第一个人工核反应是由卢瑟福在1919年实现的,他用α粒子(42He)轰击氮,产生了以下反应:
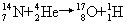
现在利用各种加速器和原子反应堆,已经实现了上万种核反应,由此获得千余种放射性同位素和各种分子、超子、反质子、反中子等基本粒子。任何核反应都遵从能量、动量、质量和电荷等守恒定律。这方面的研究对于了解原子核的结构,基本粒子间的相互作用,以及探索新的能源等方面都有重大意义。
[裂变]质量较大的重核分裂为两个质量相近的中等质量核的同时,还可能放出中子的过程。因重核的核子平均结合能比中等核的核子结合能小,所以上述过程将释放出一部分结合能。裂变有自发和人工两种。前者是重核不稳定的表现,但由于半衰期长,释放功率小。例如铀238的半衰期为1016年,故其释放的能尚无实用价值。人工裂变的反应速度可以控制,所释放的原子能可以利用。原子反应堆即为一种能源设施。
[聚变]质量较小的核聚合成质量较大的核,同时放出光子的过程。因为轻核的核子平均结合能比质量较重核的核子平均结合能大,所以上述过程将释放出一部分结合能。聚变有自发和人工两种。由于参与发生聚变反应的轻核必须相互接近到核力作用范围以内,在自然条件下,如太阳等恒星内部的温度极高,轻核具有足够大的动能以克服库仑斥力使聚变得以持续进行。在人工条件下,聚变可在爆炸或高能粒子碰撞中实现。人工控制的核聚变过程尚处在研究阶段。
[热核反应]在极高温度下进行的核聚变反应。当聚变材料达到极高温度时,聚变反应可自动进行,从而得到可以利用的能量,太阳和其它恒星中进行着热核反应。氢弹爆炸是人工热核反应。受控热核反应正在研究阶段。
[元素]又称化学元素。具有相同核电荷数(即相同质子数)的同一类原子的总称。现在已经发现108种元素,其中包括22种非金属元素和86种金属元素,金属元素与非金属元素的区分是相对的,一些非金属元素也具有类金属(半金属)的性质。元素的最小单位是原子。大多数元素都有同位素,这是由于原子核内的中子数不同引起的,如氢由质量数为1、2和3的三种同位素组成。在1940年以前,元素周期表中铀以后的元素(超铀元素)是不存在的,都是以后由人工合成的放射性元素。在自然界里元素以游离态和化合态两种形式存在,但大部分是以化合态存在的。
[化合态]元素以化合物存在的形态。如水中氢元素和氧元素都是化合态的。
[游离态]元素以单质存在的形态。如氧气中的氧元素是游离态存在的。
[纯净物]由一种单质或一种化合物组成的物质。纯净物具有固定的组成、结构和性质,例如纯净的水无色透明、无味。密度是1克/厘米3,熔点0℃,沸点100℃。自然界里纯净物极少,根据生产和科学实验的要求,可按物质不同性质进行提纯。实际上绝对纯的物质是不存在的,凡含杂质的量不至于在生产和科学实验过程中发生有害影响的物质,就当作纯净物,如作半导体材料的高纯硅,其纯度为99.999999999%,也不是完全纯净的。因此,纯净物一般是指含杂质很少具有一定纯度的物质。
[混合物]由几种不同的单质或化合物通过机械混和而成的物质。混和物没有固定的组成,混和物中各成分仍保持各自原有的性质,可以利用混和物中所含各成分的不同物理性质进行分离,例如空气中所含的氮气、氧气、惰性气体等可利用它们的沸点不同而分离开来。
[单质]由同种元素组成的纯净物。单质不可能再分解成两种或两种以上的不同物质。例如氧气(O2)、磷(P4)、铁(Fe)、银(Ag)等。单质和元素是两个不同的概念,元素是具有相同核电荷数的同一类原子的总称,元素以游离态存在时为单质,有些元素可以形成多种单质,例如碳元素可以形成金刚石、石墨和无定形碳等多种单质。根据单质的不同性质,一般单质可分为金属和非金属两大类。
[同素异形体]见同素异性体条。
[同素异性体]同一种元素组成的不同性质的单质互称同素异性体。形成同素异性体原因有三类:组成分子的原子数目不同形成的同素异性体,如O2和O3;晶体中原子的排列方式不同形成的同素异性体,如金刚石和石墨;晶体中分子的排列方式不同形成的同素异性体,如斜方硫和单斜硫。其中晶体的同素异性体也称同素异形体。
[化合物]由不同种元素组成的纯净物。化合物具有固定的组成并具有一定的性质。化合物从成分上可分为有机物和无机物两大类,从构成化合物的化学键又可分为离子化合物和共价化合物两大类。
[无机物]是无机化合物的简称,通常指不含碳元素的化合物。但少数含碳的化合物,如一氧化碳、二氧化碳、碳酸盐、氰化物等也属无机物。无机物大致可以分为氧化物、碱、酸和盐四大类。
[有机物]是有机化合物的简称。指含碳元素的化合物。组成有机物的元素除碳以外,通常还含有氢、氧、氮、硫、磷、卤素等。以前,把动植物等有机体中取得的糖类、蛋白质、油脂等物质叫有机物,到19世纪20年代,科学家先后从非生物体内取得的物质合成了许多有机物,如尿素、醋酸等,从而打破了有机物只能从有机体中取得的概念,但习惯上一直沿用有机物这个名称。有机物一般难溶于水而易溶于有机溶剂;熔点低;绝大多数有机物受热容易分解,而且容易燃烧;有机物的化学反应比较慢,并常伴有副反应发生。有机物一般可以分为烃和烃的衍生物两大类,因此有机物也就是烃和烃的衍生物的总称。
[金属]由金属元素组成的单质。具有金属光泽、不透明、有延性和展性、有良好的传热性和导电性的一类物质。在金属晶体中有中性原子、阳离子和自由电子,金属具有上述性质,都与晶体中存在自由电子有关。常温下除汞外,都以固态形式存在。化学性质一般表现较强的还原性,由于金属元素原子的价电子较少,原子半径较大,在反应中容易失去价电子成为阳离子的缘故。工业上通常把金属分为黑色金属和有色金属两大类。有色金属又可分成轻金属、重金属、贵金属和稀有金属等。
[非金属]由非金属元素组成的单质。非金属一般没有金属光泽,不是电和热的良导体,没有延性和展性。在通常情况下,非金属有的是固体,有的是气体,只有溴是液体。非金属固体大多数是分子晶体,硬变小,熔点、沸点较低,但有的非金属固体属原子晶体,如金刚石、晶体硅等,它们的硬度大,熔点、沸点较高。非金属元素的原子价电子较多,原子半径较小,在化学反应中倾向于得到电子。大多数非金属既具有氧化性,又具有还原性。金属与非金属没有严格界限,位于周期表P区左上到右下对角线附近的元素,如硼、硅、锗、砷、锑、碲等既具有非金属的性质,又具有金属的性质,可把这些元素的单质称为准金属或半金属。
[惰性气体]又称稀有气体,系周期系中零族元素,包括氦(He)、氖(Ne)、氩(Ar)、氪(Kr)、氙(Xe)和放射性元素氡(Rn)等六种元素。无色、无臭、无味,微溶于水,溶解度随分子量增大而增加。均为单原子分子。通常在化学性质上表现出惰性,至今已制出氙的稳定化合物,如XeF2、XeF4等,氪也可能形成类似氙的化合物,如KrF2,但不很稳定,它仅能在-30℃时存在。其它稀有气体的化合物尚未制出。这些气体存在于大气之中,约为空气的1%(体积),主要是氩,其它的很少。可以从液态空气分馏时制得。氡是放射性元素镭(Ra)蜕变时的产物,也叫镭射气。
[黑色金属]通常是指铁、铬、锰和铁的合金(主要指钢铁)。在各种金属中,铁在地壳中分布较集中,储量较丰富,开采和冶炼较多,价格也较廉。铸铁和钢的品种和规格很多,它们是工业上最广泛应用的金属材料,在国民经济中占有极重要的地位。铬与锰主要应用于制合金钢。铁、铬、锰及其合金都不是黑色的,而钢铁表面经常覆盖着一层黑色的四氧化三铁。这样分类,主要是从钢铁在国民经济中的重要地位出发的。
[有色金属]通常是指除铁、铬、锰和铁的合金以外的其它金属。可分为四类:⑴重金属,如铜、锌、铅、镍等;⑵轻金属,如钠、钙、镁、铝等;⑶贵金属,如金、银、铂、铱等;⑷稀有金属,如锗、铍、镧、铀等。
[重金属]一般是指密度在4.5克/厘米3以上的金属。例如铜、锌、钴、镍、钨、钼、锑、铋、铅、锡、汞等,过渡元素大都属于重金属。也有把密度在5克/厘米3以上的金属称为重金属的。
[轻金属]一般是指密度在4.5克/厘米3以下的金属。例如钠、钾、镁、钙、铝等。周期系中第一、二主族均为轻金属。也有把密度在5克/厘米3以下的金属称为轻金属的。
[贵金属]通常是指金、银和铂族金属(包括钌、铑、钯、锇、铱、铂)。这些金属在地壳中含量较少,不易开采,价格较贵,所以叫贵金属。这些金属对氧和其它试剂较稳定,金和银常用来制造装饰品和硬币。
[稀有金属]通常是指自然界中含量较少,比较分散的金属。它们难于从原料中提取,在工业上制备及应用较晚。稀有金属和普通金属没有严格界限,某些稀有金属比铜、汞、镉等金属还多。稀有金属在现代工业中具有重要的意义,往往把黑色金属、有色金属和稀有金属并列为三大类。稀有金属根据其在地壳中的分布状况及冶炼方法,可分为六类:⑴稀有轻金属,如锂、铷、铯、铍、钛等;⑵稀有难熔金属,如钨、钼、铌、钽、铪、钒等;⑶稀有分散金属(或称稀散金属),如镓、铟、铊、锗等;⑷稀土金属,包括钪、钇和镧系元素;⑸稀有贵金属,指铂族元素;⑹稀有放射性金属:包括钋、钫、镭、锕、钍、镤、铀以及从1940年以来陆续发现的十多种人造放射性元素。
[合金]由一种金属跟另一种或几种金属或非金属所组成的具有金属特性的物质叫合金。一般由各组分熔合成均匀的液体,再经冷凝而制得。按组成合金的元素数目的多少,有二元合金,三元合金和多元合金之分;按其结构的差异,又可分为共熔混和物、固溶体和金属互化物三类。一般说来,合金的熔点都低于组成它任何一种成分金属的熔点,例如用作电源保险丝的武德合金(由锡、铋、镉、铅四种金属按1∶4∶1∶2的质量比组成),熔点只有67℃,比组成它的四种金属的熔点都低。合金的硬度一般比组成的的各成分金属的硬度大,例如青铜的硬度比组成它的铜和锡的硬度都大。“齐”也是合金的意思,把含汞的合金常叫汞齐,例如钠和汞组成的合金叫钠汞齐。
[共熔混合物]合金的一种,当由金属组成的共熔的混和物凝固时,各组分分别结晶而形成的合金,例如铋镉合金与焊锡等。
[固溶体]由几种不同的化学组分所组成的均匀晶体。通常以一种物质为溶剂,在固态下溶有其它物质(溶质)的原子或分子而形成的晶体。在合金和硅酸盐系统中较多见。主要有两种类型,一是间隙固溶体,即由溶质原子溶于溶剂晶格中原子之间的间隙中而形成的,如碳溶于铁水形成的固溶体;另一是置换固溶体,即由溶质原子代替溶剂晶体中的原子而形成的,如铜溶于镍中形成的固溶体。以金属为主体形成的固溶体是合金的一种。
[金属互化物]是金属间化合物的简称。在一定条件下,金属之间相互化合而成的化合物。例如Cu3Al、CuZn3、Cu3Sn、Ag3Al、FeZn7等,金属互化物的组成并不固定,可以在一定范围内变动。例如Cu5Zn8中的锌的含量可在59-67%之间变动。大多数金属互化物是以金属键相结合,故组成元素的化合价无法确定。它是合金的一种,这类合金的传热和导电性能比组分金属低,有的具有很高电阻,通常硬而脆。
[半金属]又称准金属。介于金属与非金属之间的单质。如硼、硅、锗、砷、锑、硒、碲等。组成这些单质的元素在元素周期表中P区左上到右下的对角线附近。半金属一般为半导体。它们的导电性与金属不同,一般随温度的升高而增强。半金属在电气、冶金等方面有广泛的应用。
[准金属]见半金属条。
[氢化物]氢跟其它元素组成的二元化合物。例如氢化钠、硫化氢、硼烷等。
按其结构大致可分成三种类型:
⑴离子型氢化物(又称盐型氢化物),碱金属及碱土金属钙、锶、钡能跟氢气在高温下直接反应生成如NaH、CaH2等。在反应过程中氢夺取金属原子的价电子形成带1个单位负电荷的离子H-。这类氢化物都是离子晶体,具有较高的熔点,在熔融状态下能够导电,跟氯化钠相似。
⑵共价型氢化物(又称分子型氢化物),如HCl、H2S、NH3、CH4等都属于分子晶体,熔点、沸点较低。这类氢化物中有些具有酸性,如HCl、H2S,少数具有碱性,如NH3,另一些为中性,如CH4。
⑶金属型氢化物(又称间充氢化物),如氢化钯是金属钯晶格中间充入体积很小的氢原子而成的,无固定组成,金属吸氢后仍保持金属的特性,具有类似合金的结构,所以也称合金型氢化物。
[氧化物]氧元素跟其它元素形成的二元化合物。如氧化钙、二氧化硫、一氧化氮等。氧化物可分为不成盐氧化物(如一氧化碳、一氧化氮等)和成盐氧化物两类,后者又分为碱性氧化物(如氧化钙)、两性氧化物(如氧化铝)和酸性氧化物(如二氧化碳)。此外还有过氧化物、超氧化物、臭氧化物等。同一种元素往往有几种不同价态的氧化物,如SO2和SO3;FeO、Fe2O3和Fe3O4等。有时氧化物的含义更广泛,不限于含氧元素的二元化合物,如多元氧化物(如NiFe2O4)和有机氧化物(如氧化乙烯C2H4O2,即环氧乙烷)等。
[不成盐氧化物]又称惰性氧化物或中性氧化物。是既不能跟酸起反应,又不能跟碱起反应的氧化物,如CO和NO等。
[惰性氧化物]见不成盐氧化物条。
[中性氧化物]见不成盐氧化物条。
[碱性氧化物]能跟酸起反应,生成盐和水的氧化物。金属氧化物大多数是碱性氧化物,如氧化钙、氧化铜等。碱性氧化物对应水化物是碱,例如CaO对应的水化物是Ca(OH)2,CuO对应水化物是Cu(OH)2等,多数碱性氧化物不能直接跟水化合,有些碱性氧化物如CaO能跟水化合生成碱。
CaO+H2O=Ca(OH)2
碱性氧化物一般由金属直接氧化或难溶性碱、含氧酸盐受热分解制得。
[两性氧化物]既能跟酸起反应生成盐和水,又能跟碱起反应生成盐和水的氧化物。例如氧化铝、氧化锌等,某些过渡元素的中间价态的氧化物,如Cr2O3和Mn2O3等也是两性氧化物。两性氧化物对应的水化物是两性氢氧化物。
[酸性氧化物]能跟碱起反应,生成盐和水的氧化物。非金属氧化物多数是酸性氧化物,某些过渡元素的高价氧化物(如CrO3、Mn2O7等)也是酸性氧化物。酸性氧化物也叫酸酐,例如SO3叫硫酐,CO2叫碳酐。酸性氧化物对应的水化物是含氧酸,如SO3对应的水化物是H2SO4,CO2对应的水化物是H2CO3,SiO2对应的水化物是H2SiO3等。酸性氧化物多数能跟水直接化合生成含氧酸,少数酸性氧化物(如SiO2)不能直接跟水反应。酸性氧化物一般由非金属直接氧化或含氧酸、含氧酸盐受热分解制得。
[酸酐]简称酐。通常把由酸缩水而成的化合物称作酸酐。例如三氧化硫SO3为硫酐,可看作H2SO4分子缩去一个分子水而成;五氧化二磷(P2O5)为磷酐,可看作两个H3PO4分子缩去三个水分子而成;醋(酸)酐(CH3CO)2O,可以看作由两个醋酸CH3COOH分子缩去一个分子水而成。酸性氧化物也叫酸酐,但酸酐不一定是氧化物,例如醋酐。
[过氧化物]含有O22-基的氧化物。如Na2O2、BaO2、H2O2等均属于过氧化物。在O22-基中,氧原子跟氧原子以共价单键相结合,所以O22-也可以表示为[-O-O-]2-,O22-基中氧原子的氧化数为-1。过氧化物不稳定,有强氧化性。过氧化物能跟酸反应制取过氧化氢。
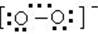 [超氧化物]含有O2-基的氧化物。如超氧化钾和超氧化铷(RbO2)等。O2-的结构如右图,含有一个未成对电子,具有顺磁性。中氧的氧化数是-1/2。超氧化物具有强氧化性,易分解放出氧气,超氧化物适用于高空飞行或其它需要携带氧气的地方。
[超氧化物]含有O2-基的氧化物。如超氧化钾和超氧化铷(RbO2)等。O2-的结构如右图,含有一个未成对电子,具有顺磁性。中氧的氧化数是-1/2。超氧化物具有强氧化性,易分解放出氧气,超氧化物适用于高空飞行或其它需要携带氧气的地方。
[臭氧化物]含有的氧化物。如臭氧化钠(NaO3)和臭氧化钾(KO3)等,只有碱金属元素能形成臭氧化物。性质极不稳定,在常温下能自动分解,是一种极强的氧化剂。
[酸]电解质电离时所生成的阳离子全部是氢离子(H+)的化合物。它能和碱或碱性氧化物反应生成盐和水,能与某些金属反应生成盐和氢气,水溶液有酸味并能使指示剂变色,如紫色石蕊变红。根据酸分子中可被金属原子置换的氢原子数目,可分为一元酸(如HCl)、二元酸(如H2SO4)、多元酸(如H3PO4)等。根据酸在水溶液中产生氢离子程度的大小(即电离度的大小),可分为强酸(如HCl、H2SO4、HNO3、HClO4等)和弱酸(如H2CO3、H2S等)。根据酸根的组成成分又可分为含氧酸(如H2SO4)和无氧酸(如HCl)。此外,根据酸碱的质子理论和电子理论,还有质子酸和路易斯酸等,因而使得“酸”这个概念包括的范围极为广泛,而且又赋予它新的含义。
[酸根]酸分子里,除去在水溶液中能够电离生成的氢离子,余下的部分称为酸根。例如盐酸根(Cl-)、硫酸根(SO42-)、磷酸根(PO43-)、碳酸根(CO32-)和碳酸氢根(HCO3-)等。酸根离子所带的负电荷数等于电离时产生氢离子的数目。
[一元酸]见酸条。
[二元酸]见酸条。
[多元酸]见酸条。
[含氧酸]见酸条。
[无氧酸]见酸条。
[多酸]亦称聚多酸或缩多酸。两个以上的酸分子缩去水分子而成的酸,也可以看作是含有两个或多个酸酐的酸。含有相同的酸酐时称为同多酸(如焦硫酸H2S2O7),对应的盐称为同多酸盐(如焦硫酸钠Na2S2O7)。元素周期表中的ⅤB和ⅥB族元素是以最容易形成多酸为其特征的。含有不同的酸酐时称为杂多酸,如12-钨磷酸H3[P(W12O40)]·xH2O,对应的盐称为杂多酸盐,如12-钨磷酸钠Na3[P(W12O40)]。
[过氧酸]简称过酸。分子中含有过氧基(-O-O-)的酸。例如过硫酸H2SO5和过二硫酸H2S2O8等。过氧酸可以看成是H2O2中的氢原子被酸根取代的产物,有如下的衍生关系。
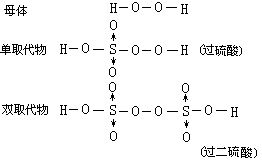
[碱]电解质电离时所生成的阴离子全部是氢氧根离子(OH-)的化合物。例如NaOH、Ca(OH)2、NH3·H2O等。能跟酸性氧化物或酸反应生成盐和水,碱的水溶液有涩味,可使紫色石蕊试液变蓝。根据碱的电离程度,可以分为强碱[如NaOH、Ca(OH)2]和弱碱(如NH3·H2O)。根据碱电离时产生的氢氧根离子的数目可分为一元碱)如NaOH)、二元碱[如Ca(OH)2]、多元碱[如Fe(OH)3]等。此外,根据酸碱的质子理论和电子理论,还有质子碱和路易斯碱,因而使得“碱”这个概念包括的范围极为广泛,而且又赋予它新的含义。
[一元碱]见碱条。
[二元碱]见碱条。
[多元碱]见碱条。
[苛性碱]碱金属氢氧化物的总称。一般指的是苛性钠(NaOH)和苛性钾(KOH)。由于它们对皮肤、羊毛、纸张、木材等具有强烈的腐蚀性而得名。
[盐]由金属离子(包括NH4+)和酸根阴离子组成的化合物。例如NaCl、(NH4)2SO4、Cu2(OH)2CO3、KHCO3等。在常温时,盐一般为晶体。它们在水中的溶解性各不相同,钾盐、钠盐、硝酸盐和铵盐很容易溶解;有些盐很难溶解,如AgCl和BaSO4等。铵盐和碳酸盐、硝酸盐、亚硝酸盐、重铬酸盐、高锰酸盐等含氧酸的盐稳定性较差,受热时可以分解。大多数盐是强电解质,其水溶液或熔液能够导电。但有些盐(如HgCl2)的水溶液几乎不导电,它在水溶液中主要以分子状态存在。根据组成的不同,盐可以分为无氧酸盐、含氧酸盐、正盐、酸式盐、碱式盐、复盐、络盐等。
[正盐]仅由金属离子(包括NH4+)和酸根阴离子组成的盐。组成中既不含可以电离的氢原子,又不含氢氧根离子。正盐可以看作酸与碱完全中和的产物。但正盐的水溶液不一定都是中性的,如果该盐可以水解,溶液一般不显中性,例如Na2CO3溶液呈碱性,(NH4)2SO4溶液呈酸性。
[酸式盐]由金属阳离子和含有可以电离的氢原子的酸根阴离子所组成的盐。例如NaHCO3、NaH2PO4等。酸式盐可以看作是多元酸中能电离的氢原子部分被中和的产物。酸式盐一般可由多元酸跟适量的碱或盐反应而得到,例如:
NaOH+H2S=NaHS+H2O
NaCl+H2SO4(浓)=NaHSO4+HCl↑
某些盐水解也可以生成酸式盐,例如:
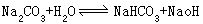
酸式盐的水溶液不一定显酸性,如NaHCO3、Na2HPO4的水溶液呈碱性。
[碱式盐]除金属阳离子和酸根阴离子外,还含有一个或几个氢氧根的盐。例如Cu2(OH)2CO3、Mg(OH)Cl等。碱式盐可看作是多元碱中的氢氧根部分被中和的产物。有些碱式盐脱水后的产物也可称为碱式盐,如碱式氯化铋(BiO)Cl、碱式碳酸铋(BiO)2CO3等。碱式盐可以由多元碱跟适量酸反应来制取,例如,
Ba(OH)2+HCl=Ba(OH)Cl+H2O
某些盐水解也可以产生碱式盐,例如,
SnCl2+H2O=Sn(OH)Cl↓+HCl
[复盐]又称重盐。两种或两种以上的不同的盐类所组成的晶形化合物。例如,明矾[KAl(SO4)2·12H2O]、硫酸亚铁铵[(NH4)2Fe(SO4)2·6H2O]等。复盐溶于水电离出的离子跟组成它简单盐所电离出的离子相同。例如:
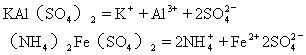
[重盐]见复盐条。
[氢氧化物]元素跟氢氧根形成的无机物叫氢氧化物。也可以看作是碱性氧化物、两性氧化物和酸性氧化物所对应的水化物。化学通式是M(OH)n。通常系指金属氢氧化物。碱性氧化物对应的氢氧化物是碱,如NaOH、Cu(OH)2等,酸性氧化物对应的氢氧化物是酸,如H2SO4、H2CO3等,两性氧化物对应的氢氧化物是两性氢氧化物,如Al(OH)3、Zn(OH)2等。当元素M的化合价(+n)较高时,氧化物的水化物易脱去一部分水而变成含水较少的化合物,例如硫酸不是H6SO6而是脱去两个水分子后的H2SO4。M(OH)n型化合物在水溶液中有酸式电离和碱式电离两种电离形式,一般金属元素的氢氧化物进行碱式电离呈碱性;非金属元素的氢氧化物进行酸式电离呈酸性。还有一些元素的氢氧化物既可以进行酸式电离,又可以进行碱式电离,这类氢氧化物称为两性氢氧化物,例如Al(OH)3、Zn(OH)2等。它们遇强酸时发生碱式电离呈碱性,遇强碱时进行酸式电离呈酸性,Al(OH)3的两种电离形式如下:

[两性氢氧化物]见氢氧化物条。
[元素符号]国际上统一采用的表示元素的化学符号,一般用元素拉丁文名称的第一个字母(大写)来表示,如:氢H、氧O。有些元素的拉丁文名称第一个字母相同,则在第一个字母后加上另一个字母(小写字)以示区别,例如:钙Ca、铜Cu、氯Cl。自人工合成104、105号元素后,由于元素的命名未取得国际上的公认,为统一起见,有关国际会议建议104号以后的新元素按原子序数的拉丁文数字命名,104、105、106、107号元素分别以Unq、Unp、Unh、Uns表示。元素符号除表示元素外,还有量的含义,即表示该元素的一个原子、表示该元素的原子量,例如O,表示氧元素,也表示氧元素的一个原子,又表示它的原子量为15.9994。
[化学式]用元素符号表示纯净物质组成的式子。化学式不仅表示该物质由哪些元素组成,还表示其中各元素原子数目的相对比数,利用化学式还可以计算出式量。化学式有实验式(最简式)、分子式、结构式、示性式、电子式等。
[实验式]又称最简式,是化学式的一种。用元素符号表示化合物分子中元素的种类和各元素原子个数的最简整数比的式子。在有机化合物中经常会出现不同的化合物具有相同的实验式,例如乙炔(C2H2)和苯(C6H6)的实验式都是CH,甲醛(CH2O)和乙酸(C2H4O2)的实验式都是CH2O等。已知化合物的最简式和分子量,就可以求出它的分子式。有些物质的实验式就是它的分子式,如甲醛CH2O和水H2O等。离子化合物晶体通常不是以分子状态存在,在实际应用上就用实验式来表示这类物质中各元素原子数目的比例关系,如NaCl表示氯化钠晶体中钠离子与氧离子数之比是1∶1。
[最简式]见实验式条。
[分子式]化学式的一种,用元素符号表示单质或化合物分子组成的式子。例如分子式H2O表示水是由氢元素和氧元素组成,表示水的一个分子,表示水分子中含有二个氢原子和一个氧原子,也表示水的分子量为18.015。分子式是通过实验测定物质的组成和分子量后求得的,一种物质只有一个分子式。不以分子形式存在的离子化合物,只表示该化合物的组成,而不是分子式。例如氯化钠晶体中只有钠离子和氯离子,不存在氯化钠分子,所以NaCl只表示氯化钠的组成,是氯化钠的化学式,而不是分子式。分子式可以和实验式相同,也可以是实验式的整数倍,例如过氧化氢的分子式是H2O2,而实验式是HO。
[结构式]化学式的一种。用元素符号通过价键相互连接表示物质分子中原子的排列顺序和结合方式的式子。例如甲烷(CH4)和乙酸(C2H4O2)的结构式分别是:
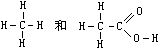
其中一条短线表示一个共价键,结构式是表示物质分子的化学结构的式子,并不表示分子的空间结构,例如甲烷分子的空间结构是正四面体。有机化合物中,同分异构现象比较普遍,例如分子式同为C2H4O2的乙酸和甲酸甲酯,它们的性质迥然不同,这是由于它们结构不同的缘故,用分子式不能区别分子组成相同,而结构相异的物质,因此,在这种情况下,使用结构式更显得重要。

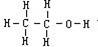 [示性式]化学式的一种,表示含有官能团的化合物分子的一种简化结构式。例如乙醇的结构式如图所示,但它的示性式是CH3CH2OH,表示乙醇分子中含有一个乙基CH3CH2-和一个羟基-OH。示性式能够明确而简便地表述同分异构体。例如乙醇和二甲醚的分子式都是C2H6O,它们的示性式分别为CH3CH2OH和CH3OCH3。
[示性式]化学式的一种,表示含有官能团的化合物分子的一种简化结构式。例如乙醇的结构式如图所示,但它的示性式是CH3CH2OH,表示乙醇分子中含有一个乙基CH3CH2-和一个羟基-OH。示性式能够明确而简便地表述同分异构体。例如乙醇和二甲醚的分子式都是C2H6O,它们的示性式分别为CH3CH2OH和CH3OCH3。
有些书籍中把CH3CH2OH叫乙醇的结构简式,而把除官能团外的其它各原子都分别合在一起写的式子叫示性式,如C2H5OH叫乙醇的示性式。结构简式和示性式都是简化的结构式,简化之后仍保持结构的特点,因此,一般书籍中把结构简式和示性式统称为示性式,不提结构简式。
[电子式]用元素符号表示原子核和内层电子,并在元素符号的周围用小黑点“·”或“×”表示原子或离子最外层电子数的图式叫电子式。例如,钠原子、氯原子、氯化钠、氯化氢的电子式分别如下:

在离子化合物的电子式中,离子的电荷用“+”或“-”表示,阴离子需加上方括号,阳离子失去外层电子后只标明电荷数即可。
用电子式可以比较简便和形象地表示离子化合物和共价化合物的形成过程。例如,MgCl2的形成过程是:
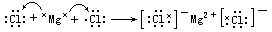
HBr的形成过程是:
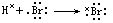
电子式只适用于某些在形成共价分子或离子化合物时,其最外电子层能达到惰性元素原子的电子层结构的原子。
 [原子结构示意图]又叫原子结构简图。用以表示原子的核电荷数和核外电子在各电子层上的排布的图式。例如,硫的原子结构示意图如右图,其中,圆圈及其中数字表示钠原子核及核内有16个质子,弧线表示电子层(硫有3个电子层),弧线上的数字表示该电子层的电子数(硫原子K层有2个电子,L层有8个电子,M层有6个电子)。原子结构示意图比较直观,易为初学者接受。但不能把弧线看作原子核外电子运行的固定轨道。
[原子结构示意图]又叫原子结构简图。用以表示原子的核电荷数和核外电子在各电子层上的排布的图式。例如,硫的原子结构示意图如右图,其中,圆圈及其中数字表示钠原子核及核内有16个质子,弧线表示电子层(硫有3个电子层),弧线上的数字表示该电子层的电子数(硫原子K层有2个电子,L层有8个电子,M层有6个电子)。原子结构示意图比较直观,易为初学者接受。但不能把弧线看作原子核外电子运行的固定轨道。
[原子结构简图]见原子结构示意图条。
[组成]化合物或混和物中各个组分的相对含量称为组成。常用质量比或质量百分数(百分组成)表示。例如水中氢和氧的质量比是1∶8,百分组成是含氢11%,含氧89%。
[定组成定律]又称定比定律。任何纯净的化合物中各元素的质量比是固定的,或者说,它们具有固定的组成。例如水,不管它是从什么地方取得的,或是用什么方法制取的,只要是纯水,水中氢元素和氧元素的质量比一定是1∶8。定组成定律是由法国化学家普鲁斯特在18世纪末根据对多种化合物的定量测定而确立的,是近代化学发展的基础。这个定律对绝大多数化合物是适用的,也有一些例外情况,如金属互化物的组成在某一定范围以内是可以改变的。
[定比定律]见定组成定律条。
[倍比定律]当甲乙两种元素互相化合形成两种或两种以上的化合物时,则在这些化合物中,与一定质量的甲元素相化合的乙元素的质量,必互成简单的整数比。例如H2O和H2O2这两种化合物都是由氢元素和氧元素组成的,H2O中氢和氧的质量比是1∶8,H2O2中氢和氧的质量比是1∶16。在这两种化合物中跟等质量的氢化合的氧的质量比为1∶2,是一个简单的整数比。这个定律是英国化学家道尔顿于1803年提出的。
[质量守恒定律]又叫物质不灭定律。参加化学反应的各物质质量总和,等于反应后生成的各物质质量总和。也就是在化学反应中,反应物的总质量等于生成物的总质量。由于化学反应是化学键的破裂与重建的过程,反应前后的原子的种类与数量都没有改变,所以化学反应前后各物质的质量总和应是相等的。这个定律是俄国科学家罗蒙诺索夫于1765年首先提出的,但是这一发现在当时并没有引起科学家的注意,直到1777年法国化学家拉瓦锡用多次实验进一步加以证实后,这一定律才获得公认。物质在发生化学反应的同时,必定伴随着能量的吸收或释放,1905年美国物理学家爱因斯坦提出质量和能量的联系公式:E=mc2,式中E为能量,m为质量,c为真空中的光速。根据这一公式可知,当一种物质的能量发生变化时,它的质量也相应地发生变化,因而质量守恒定律得到了新的发展。由于c2是一个很大的数值,只要损耗很小的质量就能释放极大的能量,在化学反应中所释放的能量不太大,很难觉察到质量的亏损,所以在化学反应中可以不加考虑。而对释放能量特别大的核反应中,就会感到质量的亏损。
[物质不灭定律]见质量守恒定律条。
[化学方程式]又称化学反应式。用化学式表示化学反应的式子。反应物写在式子的左边,生成物写在式子的右边,中间用等号连接,在特定的条件下进行的反应,可把条件写在等号处,生成物是气体或沉淀的可用“↑”或“↓”表示,例如碳酸氢钠加热时分解生成碳酸钠、水和二氧化碳,可写作
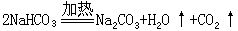
书写化学方程式必须遵循质量守恒定律,因此要在化学式前面配上适当的系数,使等号左右各种原子的总数都相等,也就是书写化学方程式时必须配平。化学方程式不仅表示参加反应的反应物和生成物的种类,而且表示反应物和生成物之间质量、物质的量的关系,对于气体物质,还能表示它们在同温同压下的体积关系,例如
N2+ 3H2=====2NH3
物质的量之比 1 ∶ 3 ∶ 2
质量比 28 ∶ 6 ∶ 34
体积比 1 ∶ 3 ∶ 2
在工农业生产和科学实验上,根据化学方程式可以进行各种计算。
[化学反应式]见化学方程式条。
[热化学方程式]标出热效应的化学方程式。把反应热写在生成物的右边,放出的热量用正值,吸收的热量用负值表示。
在写热化学方程式时:
⑴要注明反应时的温度和压力,如不注明,即表示通常状况;
⑵必须在化学式的右下侧注明物质的聚集状态,可分别用小写的s、l、g三个英文字母表示固、液、气态;
⑶化学式前的系数只表示物质的量(摩尔),不表示分子数,因此必要时可以用分数。
例如,在25℃和101325帕斯卡(1大气压)下,1摩尔氢气与1/2摩尔氧气化合生成1摩尔的水蒸气或液态水时的热化学方程式分别是:
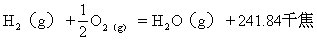
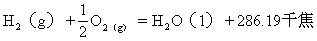
因物质的聚集态不同,因此反应热也不同。
[离子方程式]用实际参加反应的离子符号来表示离子反应的式子。例如硝酸银溶液和氯化钠溶液之间反应的离子方程式是:
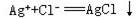
它可表示所有的可溶性银盐和盐酸或可溶性氯化物之间的反应。离子方程式可表示同一类型的离子反应,揭示了反应的实质。在离子方程式中,反应物和生成物中若为可溶性强电解质溶液,均写成离子符号;若为弱电解质(包括水)、难溶物质或气态物质时,一律写成分子式(化学式)。非溶液中的离子反应一般不用离子方程式表示。
[电离方程式]表示电解质在溶液中或受热熔化时电离成自由移动离子的式子。强电解质在溶液中完全电离,用“=”表示,弱电解质在溶液中只是部分电离,并在一定条件下达到电离平衡,所以用可逆符号表示。例如氢氧化钠和醋酸在溶液中电离方程式分别是:
NaOH=Na++OH-
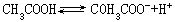
硫酸是强电解质,一级电离比较完全,二级电离就不完全(K2=1.2×10-2),所以硫酸的电离方程式一般表示为:
H2SO4=H++HSO4-
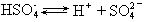
[电极反应式]表示原电池或电解池中电极上发生的氧化或还原反应的式子。
例如,铜锌原电池中的电极反应式是:
负极 Zn-2e=Zn2+
正极 Cu2++2e=Cu
氯化铜溶液在电解池中的电极反应式是:
阳极 2Cl――2e=Cl2↑
阴极 Cu2++2e=Cu
由于氧化-还原反应是同时进行的,所以电极上进行的反应又称为半反应。
[原子量]元素的原子量是一种相对质量。一种元素的原子量是该元素1摩尔质量同核素126C1摩尔质量的1/12的比值。绝大多数元素都含有两种或两种以上的同位素,因而各元素的原子量为其同位素原子质量的平均值的1摩尔质量同核素126C1摩尔质量的1/12的比值。过去化学上长期采用天然氧等于16作为原子量的标准。1929年发现自然界的氧有168O、178O、188O三种同位素,因此用天然氧等于16作为原子量的标准不够完善,物理上随后改用168O等于16作为原子量标准。
由于标准不同,容易引起混乱。1961年8月国际上正式决定采用的质量的1/12作为原子量的标准,新的原子量跟原化学原子量相差很小,例如氧的原子量从16.0000改为15.9994,相差不到十万分之四。
[同位素量]各种元素的同位素以碳元素的同位素126C原子质量的1/12为1个原子质量单位(μ)做标准的相对质量叫做同位素量。例如元素氖的三种同位素以126C作标准,它们的同位素量分别为19.99μ、20.99μ、21.99μ,元素按其所含的各种同位素的百分组成可以算出平均同位素量,上述三种同位素的百分组成分别为 90.92%,0.25%,8.83%,则氖的平均同位素量=19.99μ×90.92%+20.99μ×0.25%+21.99μ×8.83%=20.16μ。
[丰度]元素在地壳中含量的百分数,称为元素丰度,常简称丰度或克拉克值。丰度有两种表示方法,分别是质量百分数(Cw)和原子百分数(CA)。例如丰度最大的氧元素Cw为49.13%,CA为53.59%。丰度的概念有时也用来表示某元素的各种同位素的相对百分数,例如氖的三种同位素20Ne、21Ne、22Ne的丰度分别为90.92%、0.25%、8.83%,表示元素的同位素相对丰度时指的是原子百分数(CA)。
[质量数]原子核内质子数和中子数的总和。质量数是接近同位素量的整数。例如氯的两种同位素质量分别是34.969μ和36.966μ,35和37分别是这两种同位素的质量数。
[分子量]单质或化合物分子的相对质量,等于分子中各原子的原子量的总和。例如氧气(O2)的分子量为15.9994×2=31.9988,水(H2O)的分子量为1.0079×2+15.9994=18.0152。离子晶体、原子晶体和金属晶体所构成的物质,它们一般不存在单个的分子,所以也无所谓分子量。但可以用它们的化学式来计算它们的式量,例如二氧化硅(SiO2)的式量是28.0855+15.9994×2=60.0843。
[式量]物质的实验式中各原子的原子量总和。若实验式等于分子式,则式量等于分子量,例如硫酸的实验式与分子式相同,都是H2SO4,它的式量(98)就等于其分子量;若实验式与分子式不一致,则式量不等于分子量,例如葡萄糖的实验式是CH2O,分子式是C6H12O6,则式量为30,而分子量是180。
[国际单位制]简称SI制,是1960年第十一届国际计量大会建议采用的一种单位制。从1969-1975年国际标准化组织和国际计量大会经过修订、补充,正式推荐使用。国际单位制是以米、公斤、秒、安培、开尔文、摩尔、坎德拉为基本单位,其它单位均由这七个单位导出,各基本单位规定如下:
⑴米等于氪-86原子的2p6和5d5能级之间跃迁所对应的辐射在真空中的1650763.73个波长的长度;
⑵千克(公斤)是质量单位,等于国际千克原器的质量;
⑶秒是铯-133原子基态的两个超精细能级之间跃迁所对应的辐射的9192631770个周期的持续时间;
⑷安培是一恒定电流,若保持在处于真空中相距1米的两个无限长而截面可忽略的平行直导线内,则此两导线之间产生的力在每米长度上等于2×10-7牛顿,则此恒定电流的强度为1安培;
⑸开尔文是热力学温度单位,是水的三相点热力学温度的1/273.16;
⑹摩尔是一种系统的物质的量,该系统中所含的基本单元数与0.012千克碳-12中的原子数相等。在使用摩尔时,基本单元应予指明,基本单元可以是原子、分子、离子、电子及其它粒子,或者是这些粒子的特定组合;
⑺坎德拉是在101325牛顿每平方米的压强下,处于铂凝固温度的黑体的1/600000平方米表面垂直方向上的光强度。
[物质的量]是国际单位制规定的七个物理量之一,物质的量是指一定量的物质中所含微粒的多少,它的单位是摩尔。使用时要注意物质的量与摩尔数的区别,摩尔数只有数的意义,没有单位;物质的量既有数的意义又有单位。例如5摩尔水,其中5摩尔是物质的量,5是摩尔数。
[摩尔]1971年在第十四届国际计量大会上通过,把它作为国际单位制(SI)七个基本单位之一。是表示物质的量的单位。它是一种系统的物质的量,该系统中所含的基本单元数与0.012千克碳-12中的原子数相等。0.012千克碳-12中含有阿佛加德罗常数(6.02×1023)个碳原子,称为1摩尔。在使用摩尔时,应指明它所指的基本单元是原子、分子、离子、电子及其它粒子,或者是这些粒子的特定组合。
[摩尔质量]1摩尔任何物质的质量。它的单位是克/摩尔。原子的摩尔质量等于用克/摩尔表示的在数值上和该元素的原子量相等的质量;某种分子的摩尔质量等于用克/摩尔表示的在数值上和该物质的分子量相等的质量。因此摩尔质量表示1摩尔的任何物质(原子、分子、离子、电子以及其它粒子或是这些粒子的特定组合)以克/摩尔为单位的质量。
[摩尔分数]混和物或溶液中的一种物质的摩尔数与各组分的总摩尔数之比,即为该组分的摩尔分数。可以指混和物中某一组分的摩尔数与混和物总摩尔数之比;也可以指溶液中溶质的摩尔数与溶质溶剂总摩尔数之比;经常用于气体混和物中某气体的摩尔数与混和气体中各组分的总摩尔数之比。
[摩尔百分数]用百分数表示的摩尔分数。例如,混和气中某气体的摩尔数占混和气体总摩尔数的1/5,摩尔分数为1/5,摩尔百分数为20%。
[气体摩尔体积]1摩尔的任何气体在标准状况下所占的体积大约都是22.4升。这个体积称为气体摩尔体积。22.4升是一个近似数,“理想气体”的摩尔体积是22.4升,多数气体1摩尔在标准状况下所占的体积约为22.4升。
[平均分子量]22.4升气态混和物在标准状况下质量的克数,称作该混和气体的平均分子量。例如,22.4升空气在标准状况下的质量约为29克,则空气的平均分子量为29。在标准状况下22.4升的任何气态物质中所含的分子数都是阿佛加德罗常数(6.02×1023)个,因此 1摩尔混和气体的摩尔质量数,即为该混和气的平均分子量。
[阿佛加德罗定律]在同一温度和同一压强条件下,相同体积的任何气体中都含有相同数目的分子。这个结论是意大利化学家阿佛加德罗于1811年首先提出,后来为许多实验所证实的。它用于某些计算,例如气态物质的分子量计算等。根据阿佛加德罗定律,从某些气态物质在反应中体积的变化,可以推算出气态单质分子中的原子数。
[阿佛加德罗常数]1摩尔微粒(可以是分子、原子、离子、电子及其它微粒等)数经实验测得比较精确的数值是6.022045×1023个/摩尔。测定阿佛加德罗常数的方法很多,例如:由含Ag+的溶液中析出1摩尔银,需通过96484.56库仑的电量,已知每个电子的电量是1.602189×10-19库仑,因此可以得出阿佛加德罗数是
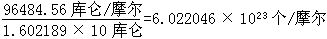
[当量]物质相互反应时,彼此所需相当的量。元素的当量是元素在化学反应中跟1.008份质量的氢或8.000份质量的氧相当的量。酸、碱、盐、氧化剂和还原剂的当量分别为:


一种物质在不同的反应中,往往有不同的当量。例如,H2SO4跟NaOH反应,生成Na2SO4时,H2SO4的当量为49,如果生成NaHSO4时,H2SO4的当量为98。
[克当量]是衡量物质质量的一种化学单位,一定量元素或化合物,用克做单位,在数值上等于当量时,这一定量物质称为一克当量。例如,氢的克当量是1.008克,氧的克当量是8.000克,盐酸的克当量是36.5克。在化学反应中,一克当量的任何物质都彼此相当。
[克当量数]表示物质克当量的数量。例如,80克氢氧化钠的克当量数为2,32克氧气的克当量数是4。
[当量定律]在任何化学反应中,反应物之间完全反应时,它们的克当量数一定相等。例如,在H2和O2化合成H2O的反应中,氢的当量为1.008,氧的当量为8.000,因此,H2与O2化合时的质量比为1.008∶8。根据当量定律,可以计算物质在化学反应中的质量关系。当量定律是德国科学家里希特尔于1791年提出的。
[氧化数]又称氧化值或氧化态。某元素一个原子的荷电数,这种荷电数由假设把每个键中的电子指定给电负性较大的原子而求得。在离子化合物中,元素原子的氧化数就等于该原子的离子电荷数,如氯化钙,钙原子的氧化数为+2,氯原子为-1。在共价化合物中,把属于两原子的共用电子对指定给两原子中电负性较大的一个以后,在两原子留下的电荷数就是它们的氧化数。如五氧化二磷,磷原子氧化数为+5,氧原子为-2。在单质中,元素的氧化数为零。元素的氧化数的变化是区别氧化-还原反应和非氧化-还原反应的主要标志。
[化合价]又称原子价。一定数目的一种元素的原子跟一定数目的其它元素原子化合的性质。它表示各种元素的原子相互化合的数目。通常以氢的化合价等于1为标准。其它元素的化合价就是与该元素的1个原子相化合的或被该元素1个原子所置换的氢原子数。化合价有电价与共价之分。
[电价]元素在离子化合物里的化合价。电价数就是这种元素的一个原子得失电子的数目,失去电子的为正价,得到电子的为负价。例如在硫化钠中,钠为+1,硫为-2。
[共价]元素在共价化合物里的化合价。共价数就是这种元素的一个原子跟其它元素的原子形成的共用电子对的数目,所以共价没有正负之分。共价不容易从分子式推断,而要从结构分析。但是不少物质的结构尚不清楚,这就难以确定共价数。现行中学化学教材里用氧化数的概念,引用到共价化合物中元素的化合价上去。根据成键原子吸引电子对能力的差别,把共价也分成正负价,实质上是元素的氧化数。
湖北省互联网违法和不良信息举报平台 | 网上有害信息举报专区 | 电信诈骗举报专区 | 涉历史虚无主义有害信息举报专区 | 涉企侵权举报专区
违法和不良信息举报电话:027-86699610 举报邮箱:58377363@163.com